

Hélène Bergès
- Carine Satgé
- Development of this new approach.
- Construction and screening of BAC libraries
- BAC clone characterization
- Arnaud BELLEC
- Construction and screening of BAC libraries
- Sequencing and comparative analysis
Dr. Patrick Vincourt
LIPM (UMR 441-2594 INRA-CNRS)
BP 52627
Chemin de Borde Rouge - Auzeville
31326 Castanet Tolosan-
Laboratoire “ Interactions Plantes Microorganismes”, UMR 441-2594(INRA-CNRS)
INRA-CNRS
31326 CASTANET TOLOSAN
Managing Director : Pascal GAMAS
Scientist in charge of the project : Patrick VINCOURT -
UMR 1095 “ Amélioration et Santé des Plantes ”
INRA – Université Blaise Pascal
63100 CLERMONT-FERRAND
Managing Director : Gilles CHARMET
Scientist in charge of the project : Felicity VEAR - D.TOURVIEILLE -
UMR 1065 « Santé végétale »des Plantes
INRA/ENITA Bordeaux
33883 VILLENAVE-D'ORNON CEDEX
Managing Director : D. THIERRY
Scientist in charge of the project : F. DELMOTTE -
UR 1258 Centre National de Ressources Génomiques Végétales
CNRGV -INRA
31326 CASTANET TOLOSAN
Managing Director / Scientist in charge of the project : Helène BERGES

FINAPEA : Fine mapping and candidate genes at a major resistance QTL to Aphanomyces euteiches in pea
FINAPEA : The project will be associated to the federative PeaMUST national Investments for the Future Consortium.
Read more
Project coordinator:
INRA-IGEPP
Marie-Laure PILET-NAY
65 rue de Saint-Brieuc
35042 Rennes
France
Email : Marie-Laure.Pilet-Nayel@inra.fr
Project partners:
INRA- UMR Agroécologie
17 rue Sully, BP 86510
21065 DIJON Cedex
France
Email : gregoire.aubert@inra.fr
INRA-CNRGV
Hélène Berges
Email : helene.berges@inra.fr
Abstract:
The sustainable development of grain legume in cropping systems is a major challenge for providing feed and food protein and limiting nitrogen applications in crop rotations. One of the major limiting factor to the production of grain legumes, especially pea predominant in France, is the common root rot disease, due to Aphanomyces euteiches. Previous studies dissected genetics of quantitative resistance to A. euteiches in pea and identified several main QTL. The project aims to fine map and identify candidate genes underlying a major resistance QTL to A. euteiches in pea, in order to support breeding efforts for future resistant varieties and discover putative functions underlying resistance QTL which are still poorly known in plants. The project will first fine map the target resistance QTL using Near-Isogenic-Line progenies previously produced and taking advantage of extensive marker resources recently developed in pea. Then, it will identify the physical sequence and candidate genes underlying the target QTL, using BAC libraries, recent long read sequencing technologies, qRT-PCR for gene expression analysis, and taking advantage of the ongoing progress of the pea genome sequencing project. The project will be associated to the federative PeaMUST national Investments for the Future Consortium. It will gather three partners of the Plant2Pro Carnot Institute and will allow innovative research to be developed in two major thematic axis of the Institute. Results will provide original basic knowledge about candidate genes underlying quantitative disease resistance in plants. It will provide new applied knowledge and tools for breeding of pea resistant varieties, which will be a key component of an integrated control strategy against A. euteiches, in combination with other methods of plant protection.
CNRGV involvement:
Responsible: Arnaud BELLEC
The CNRGV, responsible of the task2 of the project, will construct BAC libraries of two pea genotypes one resistant and the other susceptible to Aphanomyces euteiches.
| Fund agency: This project is funding by the Plant2Pro Carnot Institute. This institute is dedicated to integrated R&D “from laboratory to field” in the area of agricultural crop production. http://www.instituts-carnot.eu/en/carnot-institute/plant2pro |
|
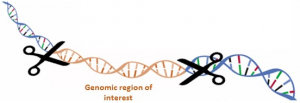
CATCH My Interest : capture of large genomic regions of interest
The CATCHMI project aims at developing a new approach to capture specific genomic regions of interest.
Read more
Project coordinator:
CNRGV
Hélène BERGES
Project partners:
INRA - LIPM
Stéphane Munos
Nicolas Langlade
24 chemin de Borde Rouge
31326 Castanet Tolosan
Email : stephane.munos@inra.fr
LAAS - CNRS
Aurélien Bancaud
7, avenue du colonnel Roche
31077 Toulouse
Email : abancaud@laas.fr
Abstract:
Agricultural research must deal with major issues on various scales, in light of the changing climatic and demographic context, where energy resources are limited. In this context of a need for improvement and adaptation of plants, genomic exploration is one of the strategic approaches of choice. Indeed, genomics will allow to define the gene content, their organization, their biological function and their variability between the different varieties. This knowledge facilitate the identification of interesting plant genes, which can play a role in biotic or abiotic resistance, in yield or in the quality process. However, the exploration of plant genome can be challenging due to the complexity of plant genomes in terms of size, repetitive elements content and various levels of ploidy.
Linking a phenotype to a genomic region is crucial to better understand biological process. However, these approaches are still based on the study of the whole genome. Most research projects require a reliable sequence of the region of interest genetically characterized or to be able to explore these regions on a larger population of individuals.
To meet these expectations, the CATCHMI project aims at developing a new approach to capture specific genomic regions of interest. This innovative strategy is based on the CRISPR / Cas9 (Clustered Regularly Interspaced Short Palindromic Repeats) technology. Indeed, this technique used in vitro can be an effective means of targeting, excising and characterizing a specific region of a genome. This CRISPR-CATCH method was tested on a bacterial genome (Jiang et al., 2015). We'll develop this approach on large and complex plant genome regions in Sunflower.
CNRGV's responsible
CNRGV involvement
| Fund agency: This project is funding by the Plant2Pro Carnot Institute. This institute is dedicated to integrated R&D “from laboratory to field” in the area of agricultural crop production. http://www.instituts-carnot.eu/en/carnot-institute/plant2pro |
|
YouTube : https://youtu.be/PWy7SZl0MW8

ZeaWall : Towards the identification of genetic determinisms involved in lignified secondary cell wall degradability through the elucidation of QTLs
ZeaWall : Towards the identification of genetic determinisms involved in lignified secondary cell wall degradability through the elucidation of QTLs
Read more
Project coordinators :
INRA URP3F
Yves Barrière
Le Chêne - RD 150
BP 80006
86600 Lusignan
Email : Yves.Barriere@lusignan.inra.fr
INRA IJPB
Matthieu Reymond
Route de Saint Cyr - RD10
78026 Versailles cedex
Email : Matthieu.Reymond@versailles.inra.fr
Project partners :
ProMaïs Crop companies (Website: http://pro-mais.org)
INRA-CNRGV
Hélène Berges
24 Chemin de Borde Rouge
31326 Castanet Tolosan
Email : helene.berges@toulouse.inra.fr
UMR de Génétique Végétale
Clémentine Vitte
Ferme du Moulon
91190 Gif-sur-Yvette, France
Email : vitte@moulon.inra.fr
Abstract :
Highlighting the genetic determinants of the lignified cell wall assembly in grasses is a major challenge for both basic research, because of the specific parietal traits of these species, and for plant breeding based on marker-assisted selection. Cell wall degradability is a limiting factor of plant energy value for cattle feeding, as well as for the production of second-generation biofuel. The research conducted, in collaboration between INRA and ProMaïs, thus aims at identifying genetic determinisms involved in cell wall related traits, taking as a model a cluster of strong effect QTLs located in a chromosome 6 genomic region of the maize recombinant inbred line (RIL) F288 x F271.
In order to characterize the region responsible for these traits, targeted sequencing of this genomic region was undertaken in the two parental lines F288 and F271 through a BAC library approach.
Publications :
Courtial A, Méchin V, Reymond M, Grima-Pettenati J, Barrière Y, 2013. Colocalizations between several QTLs for cell wall degradability and composition in the F288 x F271 early maize RIL progeny raise the question of the nature of the possible underlying determinants and breeding targets for biofuel capacity. BioEnergy Research 7(1):142-156.
Courtial A, Thomas J, Reymond M, Méchin V, Grima-Pettenati J, Barrière Y, 2013. Targeted linkage map densification to improve cell wall related QTL detection and interpretation in maize. Theor Appl Genet 126: 1151-1165.
CNRGV involvement:
Responsible: Audrey Courtial
BAC libraries construction and screening for F288 and F271 lines
BAC sequencing and comparative analysis
Controling Recombination rate for pOlyploid Crop improvement (CROC)
Controling Recombination rate for pOlyploid Crop improvement (CROC)
Read more
Meiotic recombination is a fundamental process for all sexual eukaryotes; it is required to produce balanced gametes and therefore is essential to the fertility of species. Furthermore, meiotic recombination is also crucial for plant breeding because it allows, through the formation of crossovers (COs), to reshuffle genetic material between individuals and between species. Major international efforts have been made to identify the genes that are involved in meiotic recombination in plants, primarily using diploid Arabidopsis thaliana as model system. Therefore much of this work has disregarded the consequences of polyploidy, one of the key features of crop plant genomes, on meiotic recombination.Essential questions thus remain unsolved: How is meiotic recombination regulated in polyploid (crop) species? Why and how does polyploidy increase the rate of meiotic recombination? How can such improved knowledge on recombination be exploited for crop improvement? This project will address these questions specifically, using two complementary polyploid crop species: oilseed rape (Brassica napus; AACC; 2n=38) and bread wheat (Triticum aestivum; AABBDD; 2n=42).
We will set up a complete set of integrated analyses to explore many inter-related aspects of CO regulation in polyploid crops.
Task 1 aims at characterizing the molecular underpinnings of CO suppression between homeologous chromosomes in wheat and oilseed rape. We will proceed with positional cloning of the PrBn (in oilseed rape) and Ph2 (in wheat) loci. For this latter case, particular emphasis will be placed on evaluating TaMSH7, the most promising candidate for Ph2. CROC will thus advance understanding of the mechanisms that hamper the incorporation of beneficial traits from wild relatives into crop plants by promoting a diploid-like meiosis in allopolyploids; overcoming this specific stumbling block would openthe road to the creation of new crop varieties resistant to diseases and more efficient in nitrogen use (to name only these).
Task 2 will advance understanding on the cause of the striking CO rate increase we have discovered in Brassica
digenomic triploid AAC hybrid and its possible application to wheat. We will determine whether these extra COs i) arise from one or the other CO pathways and ii) can be combined with those resulting from the mutation of an antirecombination meiotic protein. We will unravel the individual and interaction effects of three C chromosomes on the rate and distribution of COs between homologues and test whether wheat pentaploid AABBD hybrids have the same boosting effect on CO frequencies as Brassica AAC triploid hybrids. The expected outcomes will pave the way to broaden the genetic variation that is available to plant breeders.
CROC is a timely project that is shaped to address fundamental questions with practical objectives; it is directly upstream of research on innovative plant breeding technologies contributing to the competitiveness of French/European Agriculture and thus completely relevant to this call. CROC combines a group of researchers with a comprehensive and complementary expertise and set of facilities. Its strong translational emphasis ensures that the results obtained will have general significance that extends beyond oilseed rape and bread wheat. Our work will thus shed new light on the pending cause of CO variation in polyploid plant species, a critical issue for genetics, evolution and plant breeding.
ANR Project
Project coordinator:
INRA IJPB
Eric Jenczewski
Route de Saint Cyr - RD10
78026 Versailles cedex
Project partner:
INRA GDEC
Pierre Sourdille
Chemin de Beaulieu
Domaine de Crouël
63039 Clermont-Ferrand
UMR1349 IGEPP
Anne Marie Chèvre
Domaine de la Motte
INRA BP35327
35653 Le Rheu
INRA EPGV
Marie-Christine Le Paslier
rue Gaston Crémieux
CEA-IG/CNG, Bat G2, CP5724
91057 Evry
INRA-CNRGV
Hélène Berges
Chemin de Borde Rouge
31326 Castanet Tolosan
CNRGV's responsible : William Marande
CNRGV involvement:
Funded by ANR (The French National Research Agency): ANR CROC
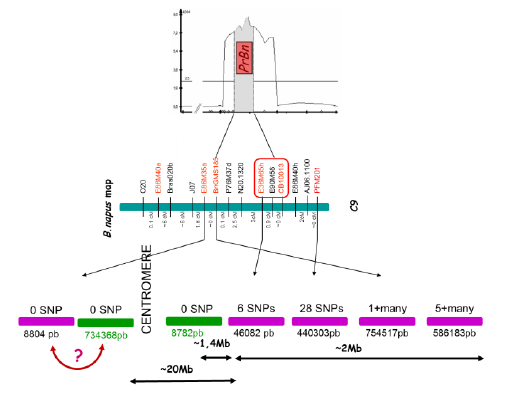
PrBn is the main determinant for CO formation between homeologous chromosomes in B. napus allohaploids; PrBn was first identified by conventional QTL analysis and mapped on chromosome C9, within a less than 10cM interval.
Using markers closely linked to PrBn as BLAST queries against the B. napus pseudomolecules, it was estimated that the entire region surrounding PrBn is ~20 Mb long and that it encompasses the centromere of C9. All but one of the markers most tightly linked to PrBn are located in an interval of ~2-3Mb on one side of the centromere that anchors 5-6 almost contiguous scaffolds. These markers are less than 2 Mb away from a meiotic recombination gene we mapped on C9.
Publication related to the project
PAGE : Plant and Animal Genome Evolution
Plant and Animal Genome Evolution (PAGE)
Read more
Project coordinator:
INRA-GDEC
Jérome SALSE
Site du Crouël
234 avenue du Brézet
63100 Clermont Ferrand
Project partners:
UR1164 URGI Unité de Recherche Génomique-Info
Hadi Quesneville
route de Saint-Cyr
78026 Versailles
INRA-LGC
Thomas Faraut
Chemin de Borde Rouge
31326 Castatnet Tolosan
CNRS-LGDP
Richard Cooke
Laboratoire Genome et Developpement des Plantes
58, Avenue P. Alduy
66860 Perpignan
DYOGEN group
CNRS UMR8541
Hugues Roest Crollius
Ecole Normale Supérieure
46 rue d’Ulm
75005 Paris
INRA-CNRGV
Hélène Berges
Chemin de Borde Rouge
31326 Castanet Tolosan
Abstract:
Given the socio-economic impact of plant and animals and the recent explosion of worldwide sequencing capacity, it is essential to develop tools to handle what will become a massive amount of information from sequenced genomes.The current proposal aims at developing tools to compare genomes to gain insight into plant and animal genome evolution since their common ancestors in terms of genome organisation (gene and repeated sequences) and the development of applicable tools for translational genomics purposes, i.e. transferring information (candidate genes, molecular markers) from sequenced genomes to non-sequenced ones, especially for genetic/physical mapping and future genome sequencing. The PAGE program will develop resources and approaches in comparative genomics and deliver public applicable tools for the scientific community.
The program involves 6 French academic partners from INRA (4 research groups) and CNRS (2 research groups) recognized through their publication records and ongoing collaborations as pioneers and major scientific groups in the field of plant and animal evolutionary genomics.
CNRGV's responsible
CNRGV involvement
Publication related to the project
PROMOSOL
|
| Durable resistance to downy mildew ( Plasmopara halstedii) in sunflower |

Read more
![]()
Durable resistance to downy mildew (Plasmopara halstedii) in sunflower
Project coordinator
Abstract
Plasmopara halstedii (downy mildew) is one of the potentially most dangerous diseases of sunflower in Europe. This disease is favoured by rainfall in spring and with the push to make earlier sowings to increase yields, the danger is likely to increase. Genetic resistance is the most efficient control method, but although new major genes giving complete resistance have been identified, after a few years use they are overcome by new races of P.halstedii. This project aims to provide tools and knowledge with the goal to draw scenarios allowing to establish strategies for a more durable resistance of sunflower to downy mildew. This will consist in developing genomic resources of P.halstedii for population genetics, studying the variability of pathogen populations and trying to determine how new races arise, and for pathotype identification ; identifying and mapping the major genes already found in large series of new sources of wild and cultivated sunflowers; fine mapping and cloning of a QTL already identified and explaining 30% of variance for a quantitative, race-non specific resistance in an RIL population, and also searching for QTL in another population to determine whether different resistance origins use different resistance mechanisms; building a Helianthus annuus genetic resource to allow more precise studies of the spatial and temporal host-parasite interaction.
Links :
http://lipm-helianthus.toulouse.inra.fr/dokuwiki/doku.php?id=start
http://www.heliagene.org/
http://www.heliagene.org/cgi/heliagene.cgi?__wb_session=WB12046189474944&__wb_main_menu=Collections&__wb_function=Collections

CNRGV involvement
Responsable : Sonia VAUTRIN
Partners

SUNYFUEL : Improving sunflower yield and quality for biofuel production by genomics and genetics
Improving sunflower yield and quality for biofuel production by genomics and genetics .
Read more
Due to its global adaptation to a wide range of southern European, water-scarce environments as well as to the introduction by breeding of a quality trait being required for biofuel production (“high oleic” type), the sunflower crop is able to take an increasing place among the environmentally safe crops dedicated to the production of raw material for the “first generation” biofuels.
This project will jointly valorise already existing genomic tools available within the international community, and wild and cultivated genetic resources of the Helianthus genus, with the aim of identifying genetic polymorphism for key genes involved in tolerance to abiotic stresses, as a target for yield improvement, and for the optimisation of harvest quality for biofuel production.
The methodological approaches will be based on association studies involving several core collections, the identification of new candidate genes from the analysis and modelling of interactions between gene expression levels and agronomic parameters describing the environmental stresses, fine mapping of QTLs already identified in previous studies, and the validation and polymorphism inventory of modifiers involved in the genetic control of oleic acid content of the oil.
![]()
![]()
Projet ANR - Réseau de Génomique Végétale - Génoplante 2010
Project coordinator :
Dr.Patrick Vincourt
LIPM (UMR 441-2594 INRA-CNRS)
BP 52627
Chemin de Borde Rouge - Auzeville
31326 Castanet Tolosan
patrick.vincourt@toulouse.inra.fr
Links :
http://lipm-helianthus.toulouse.inra.fr/dokuwiki/doku.php?id=start
http://www.heliagene.org/
http://www.heliagene.org/cgi/heliagene.cgi?__wb_session=WB12046189474944&__wb_main_menu=Collections&__wb_function=Collections
CNRGV's implications :
Responsible : Arnaud Bellec
- Construction of a Sunflower BAC library
Partners :
Laboratoire “ Interactions Plantes Microorganismes”, UMR 441-2594(INRA-CNRS)
INRA-CNRS
31326 CASTANET TOLOSAN
Managing Director : Pascal GAMAS
Scientist in charge of the project : Patrick VINCOURT
UMR 1095 “ Amélioration et Santé des Plantes ”
INRA – Université Blaise Pascal
63100 CLERMONT-FERRAND
Managing Director : Gilles CHARMET
Scientist in charge of the project : Felicity VEAR
UMR 1097 DIA-PC “ Diversité et évolution des Plantes Cultivées ”
INRA-IRD-Montpellier Sup Agro-Univ. Montpellier II
34060 MONTPELLIER
Managing Director : Serge HAMON
Scientist in charge of the project : André BERVILLE
UMR1248 Agrosystèmes et développement territorial AGIR
INRA-INPT/ENSAT
31326 CASTANET TOLOSAN
Managing Director : Michel DURU
Scientist in charge of the project : Philippe GRIEU
UMR1165 Unité de Recherche en Génomique Végétale URGV
INRA – CNRS
91057 EVRY Cedex
Managing Director : Michel CABOCHE
Scientist in charge of the project : Jean-Pierre RENOU
UR 1258 Centre National de Ressources Génomiques Végétales
CNRGV -INRA
31326 CASTANET TOLOSAN
Managing Director / Scientist in charge of the project : Helène BERGES
UR INRA 1279 “ Etude du polymorphisme des génomes végétaux ”
INRA
91 030 EVRY
Managing Director / Scientist in charge of the project : Dominique BRUNEL
UR875 Biométrie et Intelligence Artificielle
BIA -INRA
31326 CASTANET TOLOSAN
Managing Director : Roger MARTIN-CLOUAIRE
Scientist in charge of the project : Brigitte MANGIN





A wheat cysteine-rich receptor-like kinase confers broad-spectrum resistance against Septoria tritici blotch
Cyrille Saintenac, Florence Cambon, Lamia Aouini, Els Verstappen, Seyed Mahmoud Tabib Ghaffary, Théo Poucet, William Marande, Hélène Berges, Steven Xu, Maëlle Jaouannet, Bruno Favery, Julien Alassimone, Andrea Sánchez-Vallet, Justin Faris, Gert Kema, Oliver Robert & Thierry Langin
Read more
The poverty of disease resistance gene reservoirs limits the breeding of crops for durable resistance against evolutionary dynamic pathogens. Zymoseptoria tritici which causes Septoria tritici blotch (STB), represents one of the most genetically diverse and devastating wheat pathogens worldwide. No fully virulent Z. tritici isolates against synthetic wheats carrying the major resistant gene Stb16q have been identified. Here, we use comparative genomics, mutagenesis and complementation to identify Stb16q, which confers broad-spectrum resistance against Z. tritici. The Stb16q gene encodes a plasma membrane cysteine-rich receptor-like kinase that was recently introduced into cultivated wheat and which considerably slows penetration and intercellular growth of the pathogen.

















Physical Map of the Short Arm of Bread Wheat Chromosome 3D.
Journal : Plant Genome.
DOI: 10.3835/plantgenome2017.03.0021
Read more
Holušová K, Vrána J, Šafář J, Šimková H, Balcárková B, Frenkel Z, Darrier B, Paux E, Cattonaro F, Berges H, Letellier T, Alaux M, Doležel J, Bartoš J.
Journal : Plant Genome.
DOI: 10.3835/plantgenome2017.03.0021
Abstract
Bread wheat ( L.) is one of the most important crops worldwide. Although a reference genome sequence would represent a valuable resource for wheat improvement through genomics-assisted breeding and gene cloning, its generation has long been hampered by its allohexaploidy, high repeat content, and large size. As a part of a project coordinated by the International Wheat Genome Sequencing Consortium (IWGSC), a physical map of the short arm of wheat chromosome 3D (3DS) was prepared to facilitate reference genome assembly and positional gene cloning. It comprises 869 contigs with a cumulative length of 274.5 Mbp and represents 85.5% of the estimated chromosome arm size. Eighty-six Mbp of survey sequences from chromosome arm 3DS were assigned in silico to physical map contigs via next-generation sequencing of bacterial artificial chromosome pools, thus providing a high-density framework for physical map ordering along the chromosome arm. About 60% of the physical map was anchored in this single experiment. Finally, 1393 high-confidence genes were anchored to the physical map. Comparisons of gene space of the chromosome arm 3DS with genomes of closely related species [ (L.) P.Beauv., rice ( L.), and sorghum [ (L.) Moench] and homeologous wheat chromosomes provided information about gene movement on the chromosome arm.
The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution
Nature 546 : 148–152 doi:10.1038/nature22380
Read more
Hélène Badouin, Jérôme Gouzy, Christopher J. Grassa, Florent Murat, S. Evan Staton, Ludovic Cottret, Christine Lelandais-Brière, Gregory L. Owens, Sébastien Carrère, Baptiste Mayjonade, Ludovic Legrand, Navdeep Gill, Nolan C. Kane, John E. Bowers, Sariel Hubner, Arnaud Bellec, Aurélie Bérard, Hélène Bergès, Nicolas Blanchet, Marie-Claude Boniface, Dominique Brunel, Olivier Catrice, Nadia Chaidir, Clotilde Claudel, Cécile Donnadieu, Thomas Faraut, Ghislain Fievet, Nicolas Helmstetter, Matthew King, Steven J. Knapp, Zhao Lai, Marie-Christine Le Paslier, Yannick Lippi, Lolita Lorenzon, Jennifer R. Mandel, Gwenola Marage, Gwenaëlle Marchand, Elodie Marquand, Emmanuelle Bret-Mestries, Evan Morien, Savithri Nambeesan, Thuy Nguyen, Prune Pegot-Espagnet, Nicolas Pouilly, Frances Raftis, Erika Sallet, Thomas Schiex, Justine Thomas, Céline Vandecasteele, Didier Varès, Felicity Vear, Sonia Vautrin, Martin Crespi, Brigitte Mangin, John M. Burke, Jérôme Salse, Stéphane Muños, Patrick Vincourt, Loren H. Rieseberg & Nicolas B. Langlade
Abstract
The domesticated sunflower, Helianthus annuus L., is a global oil crop that has promise for climate change adaptation, because it can maintain stable yields across a wide variety of environmental conditions, including drought. Even greater resilience is achievable through the mining of resistance alleles from compatible wild sunflower relatives, including numerous extremophile species. Here we report a high-quality reference for the sunflower genome (3.6 gigabases), together with extensive transcriptomic data from vegetative and floral organs. The genome mostly consists of highly similar, related sequences and required single-molecule real-time sequencing technologies for successful assembly. Genome analyses enabled the reconstruction of the evolutionary history of the Asterids, further establishing the existence of a whole-genome triplication at the base of the Asterids II clade and a sunflower-specific whole-genome duplication around 29 million years ago. An integrative approach combining quantitative genetics, expression and diversity data permitted development of comprehensive gene networks for two major breeding traits, flowering time and oil metabolism, and revealed new candidate genes in these networks. We found that the genomic architecture of flowering time has been shaped by the most recent whole-genome duplication, which suggests that ancient paralogues can remain in the same regulatory networks for dozens of millions of years. This genome represents a cornerstone for future research programs aiming to exploit genetic diversity to improve biotic and abiotic stress resistance and oil production, while also considering agricultural constraints and human nutritional needs.
The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution
https://www.nature.com/nature/journal/v546/n7656/full/nature22380.html
Patterns of polymorphism at the self-incompatibility locus in 1,083 Arabidopsis thaliana genomes.
Mol Biol Evol. doi: 10.1093/molbev/msx122.
Read more
Authors :
Tsuchimatsu T, Goubet PM, Gallina S, Holl AC, Fobis-Loisy I, Bergès H, Marande W, Prat E, Meng D, Long Q, Platzer A, Nordborg M, Vekemans X, Castric V.
Mol Biol Evol. doi: 10.1093/molbev/msx122.
Abstract :
Although the transition to selfing in the model plant Arabidopsis thaliana involved the loss of the self-incompatibility (SI) system, it clearly did not occur due to the fixation of a single inactivating mutation at the locus determining the specificities of SI (the S-locus). At least three groups of divergent haplotypes (haplogroups), corresponding to ancient functional S-alleles, have been maintained at this locus, and extensive functional studies have shown that all three carry distinct inactivating mutations. However, the historical process of loss of SI is not well understood, in particular its relation with the last glaciation. Here, we took advantage of recently published genomic re-sequencing data in 1,083 Arabidopsis thaliana accessions that we combined with BAC sequencing to obtain polymorphism information for the whole S-locus region at a species-wide scale. The accessions differed by several major rearrangements including large deletions and inter-haplogroup recombinations, forming a set of haplogroups that are widely distributed throughout the native range and largely overlap geographically. 'Relict' A. thaliana accessions that directly derive from glacial refugia are polymorphic at the S-locus, suggesting that the three haplogroups were already present when glacial refugia from the last Ice Age became isolated. Inter-haplogroup recombinant haplotypes were highly frequent, and detailed analysis of recombination breakpoints suggested multiple independent origins. These findings suggest that the complete loss of SI in A. thaliana involved independent self-compatible mutants that arose prior to the last Ice Age, and experienced further rearrangements during post-glacial colonization.
Repeat-length variation in a wheat cellulose synthase-like gene is associated with altered tiller number and stem cell wall composition.
J Exp Bot. doi: 10.1093/jxb/erx051
Read more
Authors :
Hyles J, Vautrin S, Pettolino F, MacMillan C, Stachurski Z, Breen J, Berges H, Wicker T, Spielmeyer W.
J Exp Bot. doi: 10.1093/jxb/erx051
Abstract :
The tiller inhibition gene (tin) that reduces tillering in wheat (Triticum aestivum) is also associated with large spikes, increased grain weight, and thick leaves and stems. In this study, comparison of near-isogenic lines (NILs) revealed changes in stem morphology, cell wall composition, and stem strength. Microscopic analysis of stem cross-sections and chemical analysis of stem tissue indicated that cell walls in tin lines were thicker and more lignified than in free-tillering NILs. Increased lignification was associated with stronger stems in tin plants. A candidate gene for tin was identified through map-based cloning and was predicted to encode a cellulose synthase-like (Csl) protein with homology to members of the CslA clade. Dinucleotide repeat-length polymorphism in the 5'UTR region of the Csl gene was associated with tiller number in diverse wheat germplasm and linked to expression differences of Csl transcripts between NILs. We propose that regulation of Csl transcript and/or protein levels affects carbon partitioning throughout the plant, which plays a key role in the tin phenotype.
The chloroplast genome of Passiflora edulis (Passifloraceae) assembled from long sequence reads: structural organization and phylogenomic studies in Malpighiales
Front. Plant Sci. 8:334. DOI: 10.3389/fpls.2017.00334
Read more
Authors :
Cauz Santos LA, Freitas Munhoz C, Rodde N, Cauet S, Azevedo Santos A, Alves Penha H, Carnier Dornelas M, de Mello Varani A, Conde Xavier Oliveira G, Bergès H, Carneiro Vieira ML (2017)
Front. Plant Sci. 8:334. DOI: 10.3389/fpls.2017.00334
Abstract :
The family Passifloraceae consists of some 700 species classified in around 16 genera. Almost all its members belong to the genus Passiflora. In Brazil, the yellow passion fruit (Passiflora edulis) is of considerable economic importance, both for juice production and consumption as fresh fruit. The availability of chloroplast genomes (cp genomes) and their sequence comparisons has led to a better understanding of the evolutionary relationships within plant taxa. In this study, we obtained the complete nucleotide sequence of the P. edulis chloroplast genome, the first entirely sequenced in the Passifloraceae family. We determined its structure and organization, and also performed phylogenomic studies on the order Malpighiales and the Fabids clade. The P. edulis chloroplast genome is characterized by the presence of two copies of an inverted repeat sequence (IRA and IRB) of 26,154 bp, each separating a small single copy region of 13,378 bp and a large single copy (LSC) region of 85,720 bp. The annotation resulted in the identification of 105 unique genes, including 30 tRNAs, 4 rRNAs, and 71 protein coding genes. Also, 36 repetitive elements and 85 SSRs (microsatellites) were identified. The structure of the complete cp genome of P. edulis differs from that of other species because of rearrangement events detected by means of a comparison based on 22 members of the Malpighiales. The rearrangements were three inversions of 46,151, 3,765 and 1,631 bp, located in the LSC region. Phylogenomic analysis resulted in strongly supported trees, but this could also be a consequence of the limited taxonomic sampling used. Our results have provided a better understanding of the evolutionary relationships in the Malpighiales and the Fabids, confirming the potential of complete chloroplast genome sequences in inferring evolutionary relationships and the utility of long sequence reads for generating very accurate biological information.
Loss of pollen-specific phospholipase NOT LIKE DAD triggers gynogenesis in maize.
EMBO J. 2017 Mar 15;36(6):707-717. doi: 10.15252/embj.201796603.
Read more
Authors :
Gilles LM, Khaled A, Laffaire JB, Chaignon S, Gendrot G, Laplaige J, Bergès H, Beydon G, Bayle V, Barret P, Comadran J, Martinant JP, Rogowsky PM, Widiez T.
EMBO J. 2017 Mar 15;36(6):707-717. doi: 10.15252/embj.201796603.
Abstract :
Gynogenesis is an asexual mode of reproduction common to animals and plants, in which stimuli from the sperm cell trigger the development of the unfertilized egg cell into a haploid embryo. Fine mapping restricted a major maize QTL (quantitative trait locus) responsible for the aptitude of inducer lines to trigger gynogenesis to a zone containing a single gene NOT LIKE DAD (NLD) coding for a patatin-like phospholipase A. In all surveyed inducer lines, NLD carries a 4-bp insertion leading to a predicted truncated protein. This frameshift mutation is responsible for haploid induction because complementation with wild-type NLD abolishes the haploid induction capacity. Activity of the NLD promoter is restricted to mature pollen and pollen tube. The translational NLD::citrine fusion protein likely localizes to the sperm cell plasma membrane. In Arabidopsis roots, the truncated protein is no longer localized to the plasma membrane, contrary to the wild-type NLD protein. In conclusion, an intact pollen-specific phospholipase is required for successful sexual reproduction and its targeted disruption may allow establishing powerful haploid breeding tools in numerous crops
Links :
Jackson - 2017 - No sex please, we're (in)breeding
https://www.ncbi.nlm.nih.gov/pubmed/28228439
http://emboj.embopress.org/content/36/6.cover-expansion
http://emboj.embopress.org/
Comparative Analysis of WRKY Genes Potentially Involved in Salt Stress Responses in Triticum turgidum L. ssp. durum.
Front Plant Sci 7:2034. doi: 10.3389/fpls.2016.02034
Read more
Authors :
Yousfi FE, Makhloufi E, Marande W, Ghorbel AW., Bouzayen M and Bergès H
Front Plant Sci 7:2034. doi: 10.3389/fpls.2016.02034
Abstract :
WRKY transcription factors are involved in multiple aspects of plant growth, development and responses to biotic stresses. Although they have been found to play roles in regulating plant responses to environmental stresses, these roles still need to be explored, especially those pertaining to crops. Durum wheat is the second most widely produced cereal in the world. Complex, large and unsequenced genomes, in addition to a lack of genomic resources, hinder the molecular characterization of tolerance mechanisms. This paper describes the isolation and characterization of five TdWRKY genes from durum wheat (Triticum turgidum L. ssp. durum). A PCR-based screening of a T. turgidum BAC genomic library using primers within the conserved region of WRKY genes resulted in the isolation of five BAC clones. Following sequencing fully the five BACs, fine annotation through Triannot pipeline revealed 74.6% of the entire sequences as transposable elements and a 3.2% gene content with genes organized as islands within oceans of TEs. Each BAC clone harbored a TdWRKY gene. The study showed a very extensive conservation of genomic structure between TdWRKYs and their orthologs from Brachypodium, barley, and T. aestivum. The structural features of TdWRKY proteins suggested that they are novel members of the WRKY family in durum wheat. TdWRKY1/2/4, TdWRKY3, and TdWRKY5 belong to the group Ia, IIa, and IIc, respectively. Enrichment of cis-regulatory elements related to stress responses in the promoters of some TdWRKY genes indicated their potential roles in mediating plant responses to a wide variety of environmental stresses. TdWRKY genes displayed different expression patterns in response to salt stress that distinguishes two durum wheat genotypes with contrasting salt stress tolerance phenotypes. TdWRKY genes tended to react earlier with a down-regulation in sensitive genotype leaves and with an up-regulation in tolerant genotype leaves. The TdWRKY transcripts levels in roots increased in tolerant genotype compared to sensitive genotype. The present results indicate that these genes might play some functional role in the salt tolerance in durum wheat.
Long Read Sequencing Technology to Solve Complex Genomic Regions Assembly in Plants.
Journal of Next Generation Sequencing & Applications.
Read more
Authors :
Arnaud Bellec, Audrey Courtial, Stephane Cauet, Nathalie Rodde, Sonia Vautrin, Genseric Beydon, Nadege Arnal, Nadine Gautier, Joelle Fourment, Elisa Prat, William Marande, Yves Barriere and Helene Berges.
Journal of Next Generation Sequencing & Applications.
Abstract :
Background:
Numerous completed or on-going whole genome sequencing projects have highlighted the fact that obtaining a high quality genome sequence is necessary to address comparative genomics questions such as structural variations among genotypes and gain or loss of specific function. Despite the spectacular progress that has been made in sequencing technologies, obtaining accurate and reliable data is still a challenge, both at the whole genome scale and when targeting specific genomic regions. These problems are even more noticeable for complex plant genomes. Most plant genomes are known to be particularly challenging due to their size, high density of repetitive elements and various levels of ploidy. To overcome these problems, we have developed a strategy to reduce genome complexity by using the large insert BAC libraries combined with next generation sequencing technologies.
Results:
We compared two different technologies (Roche-454 and Pacific Biosciences PacBio RS II) to sequence pools of BAC clones in order to obtain the best quality sequence. We targeted nine BAC clones from different species (maize, wheat, strawberry, barley, sugarcane and sunflower) known to be complex in terms of sequence assembly. We sequenced the pools of the nine BAC clones with both technologies. We compared assembly results and highlighted differences due to the sequencing technologies used.
Conclusions:
We demonstrated that the long reads obtained with the PacBio RS II technology serve to obtain a better and more reliable assembly, notably by preventing errors due to duplicated or repetitive sequences in the same region.
Link :
A Metabolic Gene Cluster in the Wheat W1 and the Barley Cer-cqu Loci Determines β-Diketone Biosynthesis and Glaucousness.
Plant Cell. 2016 May 25. pii: tpc.00197.2016.
Read more
Authors :
Hen-Avivi S, Savin O, Racovita R, Lee WS, Adamki N, Malitsky S, Almekias-Siegl E, Levy M, Vautrin S, Bergès H, Friedlander G, Kartvelishvily E, Ben-Zvi G, Alkan N, Uauy C, Kanyuka K, Jetter R, Distelfeld A, Aharoni A.
Plant Cell. 2016 May 25. pii: tpc.00197.2016.
Abstract :
The glaucous appearance of wheat and barley plants, that is the light bluish-grey look of flag leaf, stem, and spike surfaces, results from deposition of cuticular β-diketone wax on their surfaces; this phenotype is associated with high yield, especially under drought conditions. Despite extensive genetic and biochemical characterization, the molecular genetic basis underlying the biosynthesis of β-diketones remains unclear. Here we discovered that the wheat W1 locus contains a metabolic gene cluster mediating β-diketone biosynthesis. The cluster comprises genes encoding proteins of several families including type-III polyketide synthases, hydrolases, and cytochrome P450s related to known fatty acid hydroxylases. The cluster region was identified in both genetic and physical maps of glaucous and glossy tetraploid wheat, demonstrating entirely different haplotypes in these accessions. Complementary evidence obtained through gene silencing in planta and heterologous expression in bacteria supports a model for a β-diketone biosynthesis pathway involving members of these three protein families. Mutations in homologous genes were identified in the barley eceriferum mutants defective in β-diketone biosynthesis, demonstrating a gene cluster also in the β-diketone biosynthesis Cer-cqu locus in barley. Hence, our findings open new opportunities to breed major cereal crops for surface features that impact yield and stress response.
Suppressed recombination and unique candidate genes in the divergent haplotype encoding Fhb1, a major Fusarium head blight resistance locus in wheat.
Theor Appl Genet. 2016 DOI: 10.1007/s00122-016-2727-x.
Read more
Authors :
Schweiger W, Steiner B, Vautrin S, Nussbaumer T, Siegwart G, Zamini M, Jungreithmeier F, Gratl V, Lemmens M, Mayer KF, Bergès H, Adam G, Buerstmayr H.
Theor Appl Genet. 2016 DOI: 10.1007/s00122-016-2727-x.
Abstract :
Fine mapping and sequencing revealed 28 genes in the non-recombining haplotype containing Fhb1 . Of these, only a GDSL lipase gene shows a pathogen-dependent expression pattern. Fhb1 is a prominent Fusarium head blight resistance locus of wheat, which has been successfully introgressed in adapted breeding material, where it confers a significant increase in overall resistance to the causal pathogen Fusarium graminearum and the fungal virulence factor and mycotoxin deoxynivalenol. The Fhb1 region has been resolved for the susceptible wheat reference genotype Chinese Spring, yet the causal gene itself has not been identified in resistant cultivars. Here, we report the establishment of a 1 Mb contig embracing Fhb1 in the donor line CM-82036. Sequencing revealed that the region of Fhb1 deviates from the Chinese Spring reference in DNA size and gene content, which explains the repressed recombination at the locus in the performed fine mapping. Differences in genes expression between near-isogenic lines segregating for Fhb1 challenged with F. graminearum or treated with mock were investigated in a time-course experiment by RNA sequencing. Several candidate genes were identified, including a pathogen-responsive GDSL lipase absent in susceptible lines. The sequence of the Fhb1 region, the resulting list of candidate genes, and near-diagnostic KASP markers for Fhb1 constitute a valuable resource for breeding and further studies aiming to identify the gene(s) responsible for F. graminearum and deoxynivalenol resistance.
Advancing Eucalyptus Genomics: Cytogenomics Reveals Conservation of Eucalyptus Genomes.
Front Plant Sci. 2016 Apr 22;7:510. doi: 10.3389/fpls.2016.00510
Read more
Authors :
Ribeiro T, Barrela RM, Bergès H, Marques C, Loureiro J, Morais-Cecílio L, Paiva JA.
Front Plant Sci. 2016 Apr 22;7:510. doi: 10.3389/fpls.2016.00510
Abstract :
The genus Eucalyptus encloses several species with high ecological and economic value, being the subgenus Symphyomyrtus one of the most important. Species such as E. grandis and E. globulus are well characterized at the molecular level but knowledge regarding genome and chromosome organization is very scarce. Here we characterized and compared the karyotypes of three economically important species, E. grandis, E. globulus, and E. calmadulensis, and three with ecological relevance, E. pulverulenta, E. cornuta, and E. occidentalis, through an integrative approach including genome size estimation, fluorochrome banding, rDNA FISH, and BAC landing comprising genes involved in lignin biosynthesis. All karyotypes show a high degree of conservation with pericentromeric 35S and 5S rDNA loci in the first and third pairs, respectively. GC-rich heterochromatin was restricted to the 35S rDNA locus while the AT-rich heterochromatin pattern was species-specific. The slight differences in karyotype formulas and distribution of AT-rich heterochromatin, along with genome sizes estimations, support the idea of Eucalyptus genome evolution by local expansions of heterochromatin clusters. The unusual co-localization of both rDNA with AT-rich heterochromatin was attributed mainly to the presence of silent transposable elements in those loci. The cinnamoyl CoA reductase gene (CCR1) previously assessed to linkage group 10 (LG10) was clearly localized distally at the long arm of chromosome 9 establishing an unexpected correlation between the cytogenetic chromosome 9 and the LG10. Our work is novel and contributes to the understanding of Eucalyptus genome organization which is essential to develop successful advanced breeding strategies for this genus.
The wheat Sr50 gene reveals rich diversity at a cereal disease resistance locus
Nature Plants. Article number:15186 (2015). DOI:10.1038/nplants.2015.186
Read more
Nature Plants. Article number:15186 (2015). DOI:10.1038/nplants.2015.186
Authors :
Rohit Mago, Peng Zhang, Sonia Vautrin, Hana Šimková, Urmil Bansal, Ming-Cheng Luo, Matthew Rouse, Haydar Karaoglu, Sambasivam Periyannan, James Kolmer, Yue Jin, Michael A. Ayliffe, Harbans Bariana, Robert F. Park, Robert McIntosh, Jaroslav Doležel, Hélène Bergès, Wolfgang Spielmeyer, Evans S. Lagudah, Jeff G. Ellis, Peter N. Dodds
Abstract :
We identify the wheat stem rust resistance gene Sr50 (using physical mapping, mutation and complementation) as homologous to barley Mla, encoding a coiled-coil nucleotide-binding leucine-rich repeat (CC-NB-LRR) protein. We show that Sr50 confers a unique resistance specificity different from Sr31 and other genes on rye chromosome 1RS, and is effective against the broadly virulent Ug99 race lineage. Extensive haplotype diversity at the rye Sr50 locus holds promise for mining effective resistance genes.
Link :
FRIZZY PANICLE drives supernumerary spikelets in bread wheat (T. aestivum L.).
Plant Physiol. 2015 Jan;167(1):189-99. doi: 10.1104/pp.114.250043
Read more
Authors :
Dobrovolskaya O, Pont C, Sibout R, Martinek P, Badaeva E, Murat F, Chosson A, Watanabe N, Prat E, Gautier N, Gautier V, Poncet C, Orlov Y, Krasnikov A, Bergès H, Salina E, Laikova L, Salse J.
Plant Physiol. 2015 Jan;167(1):189-99. doi: 10.1104/pp.114.250043
Abstract :
Bread wheat (Triticum aestivum) inflorescences, or spikes, are characteristically unbranched and normally bear one spikelet per rachis node. Wheat mutants on which supernumerary spikelets (SSs) develop are particularly useful resources for work towards understanding the genetic mechanisms underlying wheat inflorescence architecture and, ultimately, yield components. Here, we report the characterization of genetically unrelated mutants leading to the identification of the wheat FRIZZY PANICLE (FZP) gene, encoding a member of the APETALA2/Ethylene Response Factor transcription factor family, which drives the SS trait in bread wheat. Structural and functional characterization of the three wheat FZP homoeologous genes (WFZP) revealed that coding mutations of WFZP-D cause the SS phenotype, with the most severe effect when WFZP-D lesions are combined with a frameshift mutation in WFZP-A. We provide WFZP-based resources that may be useful for genetic manipulations with the aim of improving bread wheat yield by increasing grain number.
The first insight into the Salvia (lamiaceae) genome via BAC library construction and high-throughput sequencing of target BAC clones.
(2015). Pak. J. Bot., 47(4): 1347-1357.
Read more
Authors :
Da Cheng Hao, Sonia Vautrin, Chi Song, Ying Jie Zhu, Helene Berges, Chao Sun and Shi Lin Chen
Pak. J. Bot., 47(4): 1347-1357, 2015.
Abstract :
Salvia is a representative genus of Lamiaceae, a eudicot family with significant species diversity and population adaptibility. One of the key goals of Salvia genomics research is to identify genes of adaptive significance. This information may help to improve the conservation of adaptive genetic variation and the management of medicinal plants to increase their health and productivity. Large-insert genomic libraries are a fundamental tool for achieving this purpose. We report herein the construction, characterization and screening of a gridded BAC library for Salvia officinalis (sage). The S. officinalis BAC library consists of 17,764 clones and the average insert size is 107 Kb, corresponding to similar to 3 haploid genome equivalents. Seventeen positive clones (average insert size 115 Kb) containing five terpene synthase (TPS) genes were screened out by PCR and 12 of them were subject to Illumina HiSeq 2000 sequencing, which yielded 28,097,480 90-bp raw reads (2.53 Gb). Scaffolds containing sabinene synthase (Sab), a Sab homolog, TPS3 (kaurene synthase-like 2), copalyl diphosphate synthase 2 and one cytochrome P450 gene were retrieved via de novo assembly and annotation, which also have flanking noncoding sequences, including predicted promoters and repeat sequences. Among 2,638 repeat sequences, there are 330 amplifiable microsatellites. This BAC library provides a new resource for Lamiaceae genomic studies, including microsatellite marker development, physical mapping, comparative genomics and genome sequencing. Characterization of positive clones provided insights into the structure of the Salvia genome. These sequences will be used in the assembly of a future genome sequence for S. officinalis.
The physical map of wheat chromosome 5DS revealed gene duplications and small rearrangements.
BMC Genomics. 2015 Jun 13;16:453. doi: 10.1186/s12864-015-1641-y.
Read more
Authors :
Akpinar BA, Magni F, Yuce M, Lucas SJ, Šimková H, Šafář J, Vautrin S, Bergès H, Cattonaro F, Doležel J, Budak H.
BMC Genomics. 2015 Jun 13;16:453. doi: 10.1186/s12864-015-1641-y.
Abstract :
BACKGROUND:
The substantially large bread wheat genome, organized into highly similar three sub-genomes, renders genomic research challenging. The construction of BAC-based physical maps of individual chromosomes reduces the complexity of this allohexaploid genome, enables elucidation of gene space and evolutionary relationships, provides tools for map-based cloning, and serves as a framework for reference sequencing efforts. In this study, we constructed the first comprehensive physical map of wheat chromosome arm 5DS, thereby exploring its gene space organization and evolution.
RESULTS:
The physical map of 5DS was comprised of 164 contigs, of which 45 were organized into 21 supercontigs, covering 176 Mb with an N50 value of 2,173 kb. Fifty-eight of the contigs were larger than 1 Mb, with the largest contig spanning 6,649 kb. A total of 1,864 molecular markers were assigned to the map at a density of 10.5 markers/Mb, anchoring 100 of the 120 contigs (>5 clones) that constitute ~95 % of the cumulative length of the map. Ordering of 80 contigs along the deletion bins of chromosome arm 5DS revealed small-scale breaks in syntenic blocks. Analysis of the gene space of 5DS suggested an increasing gradient of genes organized in islands towards the telomere, with the highest gene density of 5.17 genes/Mb in the 0.67-0.78 deletion bin, 1.4 to 1.6 times that of all other bins.
CONCLUSIONS:
Here, we provide a chromosome-specific view into the organization and evolution of the D genome of bread wheat, in comparison to one of its ancestors, revealing recent genome rearrangements. The high-quality physical map constructed in this study paves the way for the assembly of a reference sequence, from which breeding efforts will greatly benefit.
Major haplotype divergence including multiple germin-like protein genes, at the wheat Sr2 adult plant stem rust resistance locus.
BMC Plant Biol. 2014. 14(1):1585.
Read more
Authors
Mago R, Tabe L, Vautrin S, Imková H, Kubaláková M, Upadhyaya N, Berges H, Kong X, Breen J, Dole El J, Appels R, Ellis J, Spielmeyer W
BMC Plant Biol. 2014. 14(1):1585.
Abstract
The adult plant stem rust resistance gene Sr2 was introgressed into hexaploid wheat cultivar (cv) Marquis from tetraploid emmer wheat cv Yaroslav, to generate stem rust resistant cv Hope in the 1920s. Subsequently, Sr2 has been widely deployed and has provided durable partial resistance to all known races of Puccinia graminis f. sp. tritici. This report describes the physical map of the Sr2-carrying region on the short arm of chromosome 3B of cv Hope and compares the Hope haplotype with non-Sr2 wheat cv Chinese Spring.Results Sr2 was located to a region of 867 kb on chromosome 3B in Hope, which corresponded to a region of 567 kb in Chinese Spring. The Hope Sr2 region carried 34 putative genes but only 17 were annotated in the comparable region of Chinese Spring. The two haplotypes differed by extensive DNA sequence polymorphisms between flanking markers as well as by a major insertion/deletion event including ten Germin-Like Protein (GLP) genes in Hope that were absent in Chinese Spring. Haplotype analysis of a limited number of wheat genotypes of interest showed that all wheat genotypes carrying Sr2 possessed the GLP cluster; while, of those lacking Sr2, some, including Marquis, possessed the cluster, while some lacked it. Thus, this region represents a common presence-absence polymorphism in wheat, with presence of the cluster not correlated with presence of Sr2. Comparison of Hope and Marquis GLP genes on 3BS found no polymorphisms in the coding regions of the ten genes but several SNPs in the shared promoter of one divergently transcribed GLP gene pair and a single SNP downstream of the transcribed region of a second GLP.ConclusionPhysical mapping and sequence comparison showed major haplotype divergence at the Sr2 locus between Hope and Chinese Spring. Candidate genes within the Sr2 region of Hope are being evaluated for the ability to confer stem rust resistance. Based on the detailed mapping and sequencing of the locus, we predict that Sr2 does not belong to the NB-LRR gene family and is not related to previously cloned, race non-specific rust resistance genes Lr34 and Yr36.
Dominance hierarchy arising from the evolution of a complex small RNA regulatory network.
Science. 346(6214):1200-5. doi: 10.1126/science.1259442.
Read more
Authors
Durand E1, Méheust R1, Soucaze M1, Goubet PM1, Gallina S1, Poux C1, Fobis-Loisy I2, Guillon E2, Gaude T2, Sarazin A3, Figeac M4, Prat E5, Marande W5, Bergès H5, Vekemans X1, Billiard S1, Castric V6.
Science. 346(6214):1200-5. doi: 10.1126/science.1259442.
Abstract
The prevention of fertilization through self-pollination (or pollination by a close relative) in the Brassicaceae plant family is determined by the genotype of the plant at the self-incompatibility locus (S locus). The many alleles at this locus exhibit a dominance hierarchy that determines which of the two allelic specificities of a heterozygous genotype is expressed at the phenotypic level. Here, we uncover the evolution of how at least 17 small RNA (sRNA)-producing loci and their multiple target sites collectively control the dominance hierarchy among alleles within the gene controlling the pollen S-locus phenotype in a self-incompatible Arabidopsis species. Selection has created a dynamic repertoire of sRNA-target interactions by jointly acting on sRNA genes and their target sites, which has resulted in a complex system of regulation among alleles.
Begin at the beginning: A BAC-end view of the passion fruit (Passiflora) genome.
BMC Genomics. 2014 Sep 26;15:816. doi: 10.1186/1471-2164-15-816
Read more
Authors
Santos AA, Penha HA, Bellec A, Munhoz Cde F, Pedrosa-Harand A, Bergès H, Vieira ML.
BMC Genomics. 2014 Sep 26;15:816. doi: 10.1186/1471-2164-15-816
Abstract
Background:
The passion fruit (Passiflora edulis) is a tropical crop of economic importance both for juice production and consumption as fresh fruit. The juice is also used in concentrate blends that are consumed worldwide. However, very little is known about the genome of the species. Therefore, improving our understanding of passion fruit genomics is essential and to some degree a pre-requisite if its genetic resources are to be used more efficiently. In this study, we have constructed a large-insert BAC library and provided the first view on the structure and content of the passion fruit genome, using BAC-end sequence (BES) data as a major resource.
Results:
The library consisted of 82,944 clones and its levels of organellar DNA were very low. The library represents six haploid genome equivalents, and the average insert size was 108 kb. To check its utility for gene isolation, successful macroarray screening experiments were carried out with probes complementary to eight Passiflora gene sequences available in public databases. BACs harbouring those genes were used in fluorescent in situ hybridizations and unique signals were detected for four BACs in three chromosomes (n = 9). Then, we explored 10,000 BES and we identified reads likely to contain repetitive mobile elements (19.6% of all BES), simple sequence repeats and putative proteins, and to estimate the GC content (~42%) of the reads. Around 9.6% of all BES were found to have high levels of similarity to plant genes and ontological terms were assigned to more than half of the sequences analysed (940). The vast majority of the top-hits made by our sequences were to Populus trichocarpa (24.8% of the total occurrences), Theobroma cacao (21.6%), Ricinus communis (14.3%), Vitis vinifera (6.5%) and Prunus persica (3.8%).
Conclusions:
We generated the first large-insert library for a member of Passifloraceae. This BAC library provides a new resource for genetic and genomic studies, as well as it represents a valuable tool for future whole genome study. Remarkably, a number of BAC-end pair sequences could be mapped to intervals of the sequenced Arabidopsis thaliana, V. vinifera and P. trichocarpa chromosomes, and putative collinear microsyntenic regions were identified.
Isolation and molecular characterization of ERF1, an ethylene response factor gene from durum wheat (Triticum turgidum L. subsp. durum), potentially involved in salt-stress responses.
J Exp Bot. 2014
Read more
Authors
Makhloufi E, Yousfi FE, Marande W, Mila I, Hanana M, Bergès H, Mzid R, Bouzayen M.
J Exp Bot. 2014
Abstract
As food crop, wheat is of prime importance for human society. Nevertheless, our understanding of the genetic and molecular mechanisms controlling wheat productivity conditions has been, so far, hampered by the lack of sufficient genomic resources. The present work describes the isolation and characterization of TdERF1, an ERF gene from durum wheat (Triticum turgidum L. subsp. durum). The structural features of TdERF1 supported the hypothesis that it is a novel member of the ERF family in durum wheat and, considering its close similarity to TaERF1 of Triticum aestivum, it probably plays a similar role in mediating responses to environmental stresses. TdERF1 displayed an expression pattern that discriminated between two durum wheat genotypes contrasted with regard to salt-stress tolerance. The high number of cis-regulatory elements related to stress responses present in the TdERF1 promoter and the ability of TdERF1 to regulate the transcription of ethylene and drought-responsive promoters clearly indicated its potential role in mediating plant responses to a wide variety of environmental constrains. TdERF1 was also regulated by abscisic acid, ethylene, auxin, and salicylic acid, suggesting that it may be at the crossroads of multiple hormone signalling pathways. Four TdERF1 allelic variants have been identified in durum wheat genome, all shown to be transcriptionally active. Interestingly, the expression of one allelic form is specific to the tolerant genotype, further supporting the hypothesis that this gene is probably associated with the susceptibility/tolerance mechanism to salt stress. In this regard, the TdERF1 gene may provide a discriminating marker between tolerant and sensitive wheat varieties.
Structural and functional partitioning of bread wheat chromosome 3B.
Science. 2014 Jul 18;345(6194):1249721. doi: 10.1126/science.1249721.
Read more
Authors
Choulet F, Alberti A, Theil S, Glover N, Barbe V, Daron J, Pingault L, Sourdille P, Couloux A, Paux E, Leroy P, Mangenot S, Guilhot N, Le Gouis J, Balfourier F, Alaux M, Jamilloux V, Poulain J, Durand C, Bellec A, Gaspin C, Safar J, Dolezel J, Rogers J, Vandepoele K, Aury JM, Mayer K, Berges H, Quesneville H, Wincker P, Feuillet C.
Science. 2014 Jul 18;345(6194):1249721. doi: 10.1126/science.1249721.
Abstract
We produced a reference sequence of the 1-gigabase chromosome 3B of hexaploid bread wheat. By sequencing 8452 bacterial artificial chromosomes in pools, we assembled a sequence of 774 megabases carrying 5326 protein-coding genes, 1938 pseudogenes, and 85% of transposable elements. The distribution of structural and functional features along the chromosome revealed partitioning correlated with meiotic recombination. Comparative analyses indicated high wheat-specific inter- and intrachromosomal gene duplication activities that are potential sources of variability for adaption. In addition to providing a better understanding of the organization, function, and evolution of a large and polyploid genome, the availability of a high-quality sequence anchored to genetic maps will accelerate the identification of genes underlying important agronomic traits.
Building the sugarcane genome for biotechnology and identifying evolutionary trends.
BMC Genomics. 2014 Jun 30;15(1):540.
Read more
Authors
De Setta N, Monteiro-Vitorello CB, Metcalfe CJ, Cruz GM, Del Bem LE, Vicentini R, Nogueira FT, Campos RA, Nunes SL, Turrini PC, Vieira AP, Ochoa Cruz EA, Corrêa TC, Hotta CT, de Mello Varani A, Vautrin S, da Trindade AS, de Mendonça Vilela M, Lembke CG, Sato PM, de Andrade RF, Nishiyama MY Jr, Cardoso-Silva CB, Scortecci KC, Garcia AA, Carneiro MS, Kim C, Paterson AH, Bergès H, D Hont A, de Souza AP, Souza GM, Vincentz M, Kitajima JP, Van Sluys MA.
BMC Genomics. 2014 Jun 30;15(1):540.
Abstract
BACKGROUND:
Sugarcane is the source of sugar in all tropical and subtropical countries and is becoming increasingly important for bio-based fuels. However, its large (10 Gb), polyploid, complex genome has hindered genome based breeding efforts. Here we release the largest and most diverse set of sugarcane genome sequences to date, as part of an on-going initiative to provide a sugarcane genomic information resource, with the ultimate goal of producing a gold standard genome.
RESULTS:
Three hundred and seventeen chiefly euchromatic BACs were sequenced. A reference set of one thousand four hundred manually-annotated protein-coding genes was generated. A small RNA collection and a RNA-seq library were used to explore expression patterns and the sRNA landscape. In the sucrose and starch metabolism pathway, 16 non-redundant enzyme-encoding genes were identified. One of the sucrose pathway genes, sucrose-6-phosphate phosphohydrolase, is duplicated in sugarcane and sorghum, but not in rice and maize. A diversity analysis of the s6pp duplication region revealed haplotype-structured sequence composition. Examination of hom(e)ologous loci indicate both sequence structural and sRNA landscape variation. A synteny analysis shows that the sugarcane genome has expanded relative to the sorghum genome, largely due to the presence of transposable elements and uncharacterized intergenic and intronic sequences.
CONCLUSIONS:
This release of sugarcane genomic sequences will advance our understanding of sugarcane genetics and contribute to the development of molecular tools for breeding purposes and gene discovery.
Sequence-Based Analysis of Structural Organization and Composition of the Cultivated Sunflower (Helianthus annuus L.) Genome.
Biology (Basel). 3(2):295-319. doi: 10.3390/biology3020295
Read more
Authors
Gill N, Buti M, Kane N, Bellec A, Helmstetter N, Berges H, Rieseberg LH.
Biology (Basel). 3(2):295-319. doi: 10.3390/biology3020295
Abstract
Sunflower is an important oilseed crop, as well as a model system for evolutionary studies, but its 3.6 gigabase genome has proven difficult to assemble, in part because of the high repeat content of its genome. Here we report on the sequencing, assembly, and analyses of 96 randomly chosen BACs from sunflower to provide additional information on the repeat content of the sunflower genome, assess how repetitive elements in the sunflower genome are organized relative to genes, and compare the genomic distribution of these repeats to that found in other food crops and model species. We also examine the expression of transposable element-related transcripts in EST databases for sunflower to determine the representation of repeats in the transcriptome and to measure their transcriptional activity. Our data confirm previous reports in suggesting that the sunflower genome is >78% repetitive. Sunflower repeats share very little similarity to other plant repeats such as those of Arabidopsis, rice, maize and wheat; overall 28% of repeats are "novel" to sunflower. The repetitive sequences appear to be randomly distributed within the sequenced BACs. Assuming the 96 BACs are representative of the genome as a whole, then approximately 5.2% of the sunflower genome comprises non TE-related genic sequence, with an average gene density of 18kbp/gene. Expression levels of these transposable elements indicate tissue specificity and differential expression in vegetative and reproductive tissues, suggesting that expressed TEs might contribute to sunflower development. The assembled BACs will also be useful for assessing the quality of several different draft assemblies of the sunflower genome and for annotating the reference sequence.
The homoeologous genes encoding chalcone-flavanone isomerase in Triticum aestivum L.: structural characterization and expression in different parts of wheat plant.
Gene. 538(2):334-41. doi: 10.1016/j.gene.2014.01.008.
Read more
Authors
Shoeva OY, Khlestkina EK, Berges H, Salina EA.
Gene. 538(2):334-41. doi: 10.1016/j.gene.2014.01.008.
Abstract
Chalcone-flavanone isomerase (CHI; EC 5.5.1.6.) participates in the early step of flavonoid biosynthesis, related to plant adaptive and protective responses to environmental stress. The bread wheat genomic sequences encoding CHI were isolated, sequenced and mapped to the terminal segment of the long arms of chromosomes 5A, 5B and 5D. The loss of the final Chi intron and junction of the two last exons was found in the wheat A, B and D genomes compared to the Chi sequences of most other plant species. Each of the three diploid genomes of hexaploid wheat encodes functional CHI; however, transcription of the three homoeologous genes is not always co-regulated. In particular, the three genes demonstrated different response to salinity in roots: Chi-D1 was up-regulated, Chi-A1 responds medially, whereas Chi-B1 was not activated at all. The observed variation in transcriptional activity between the Chi homoeologs is in a good agreement with structural diversification of their promoter sequences. In addition, the correlation between Chi transcription and anthocyanin pigmentation in different parts of wheat plant has been studied. The regulatory genes controlling anthocyanin pigmentation of culm and pericarp modulated transcription of the Chi genes. However, in other organs, there was no strong relation between tissue pigmentation and the transcription of the Chi genes, suggesting complex regulation of the Chi expression in most parts of wheat plant.
Meiotic gene evolution: can you teach a new dog new tricks?
Mol Biol Evol. 2014
Read more
Authors
Lloyd A, Ranoux M, Vautrin S, Glover N, Fourment J, Charif D, Choulet F, Lassalle G, Marande W, Tran J, Granier F, Pingault L, Remay A, Marquis C, Belcram H, Chalhoub B, Feuillet C, Bergès H, Sourdille P, Jenczewski E.
MolBiol Evol. 2014
Abstract
Meiosis, the basis of sex, evolved through iterative gene duplications. To understand whether subsequent duplications have further enriched the core meiotic "tool-kit", we investigated the fate of meiotic gene duplicates following Whole Genome Duplication (WGD), a common occurrence in eukaryotes. We show that meiotic genes return to a single copy more rapidly than genome-wide average in Angiosperms, one of the lineages in which WGD is most vividly exemplified. The rate at which duplicates are lost decreases through time, a tendency that is also observed genome-wide and may thus prove to be a general trend post-WGD. The sharpest decline is observed for the subset of genes mediating meiotic recombination; however, we found no evidence that the presence of these duplicates is counter-selected in two recent polyploid crops selected for fertility. We therefore propose that their loss is passive, highlighting how quickly WGDs are resolved in the absence of selective duplicate retention.
The physical map of wheat chromosome 1BS provides insights into its gene space organization and evolution.
Genome Biol. 2013 Dec 20;14(12):R138.
Read more
Authors
Raats D, Frenkel Z, Krugman T, Dodek I, Sela H, Imková H, Magni F, Cattonaro F, Vautrin S, Bergès H, Wicker T, Keller B, Leroy P, Philippe R, Paux E, Dole El J, Feuillet C, Korol A, Fahima T
Genome Biol. 2013 Dec 20;14(12):R138.
Abstract
BACKGROUND:
The wheat genome sequence is an essential tool for advanced genomic research and improvements. The generation of a high-quality wheat genome sequence is challenging due to its complex 17 Gb polyploid genome. To overcome these difficulties, sequencing through the construction of BAC-based physical maps of individual chromosomes is employed by the wheat genomics community. Here, we present the construction of the first comprehensive physical map of chromosome 1BS, and illustrate its unique gene space organization and evolution.
RESULTS:
Fingerprinted BAC clones were assembled into 57 long scaffolds, anchored and ordered with 2,438 markers covering 83% of chromosome 1BS. The BAC-based chromosome 1BS physical map and gene order of the orthologous regions of model grass species were consistent, providing strong support for the reliability of the chromosome 1BS assembly. Chromosome 1BS gene space spans the entire length of the chromosome arm, with 76% of the genes organized in small gene-islands, accompanied by a two fold increase in gene density from the centromere to the telomere.
CONCLUSIONS:
This study provides new evidence on common and chromosome-specific features in the organization and evolution of the wheat genome, including a non-uniform distribution of gene density along the centromere-telomere axis, abundance of non-syntenic genes, the degree of colinearity with other grass genomes, and a non-uniform size expansion along the centromere-telomere axis, compared with other model cereal genomes. The high quality physical map constructed in this study provides a solid basis for the assembly of a reference sequence of chromosome 1BS and for breeding applications.
A Physical Map of the Short Arm of Wheat Chromosome 1A.
PLoS One. 8(11):e80272. doi: 10.1371/journal.pone.0080272.
Read more
Authors
Breen J, Wicker T, Shatalina M, Frenkel Z, Bertin I, Philippe R, Spielmeyer W, Simková H, Safář J, Cattonaro F, Scalabrin S, Magni F, Vautrin S, Bergès H; International Wheat Genome Sequencing Consortium, Paux E, Fahima T, Doležel J, Korol A, Feuillet C, Keller B.
PLoS One. 8(11):e80272. doi: 10.1371/journal.pone.0080272.
Abstract
Bread wheat (Triticum aestivum) has a large and highly repetitive genome which poses major technical challenges for its study. To aid map-based cloning and future genome sequencing projects, we constructed a BAC-based physical map of the short arm of wheat chromosome 1A (1AS). From the assembly of 25,918 high information content (HICF) fingerprints from a 1AS-specific BAC library, 715 physical contigs were produced that cover almost 99% of the estimated size of the chromosome arm. The 3,414 BAC clones constituting the minimum tiling path were end-sequenced. Using a gene microarray containing ∼40 K NCBI UniGene EST clusters, PCR marker screening and BAC end sequences, we arranged 160 physical contigs (97 Mb or 35.3% of the chromosome arm) in a virtual order based on synteny with Brachypodium, rice and sorghum. BAC end sequences and information from microarray hybridisation was used to anchor 3.8 Mbp of Illumina sequences from flow-sorted chromosome 1AS to BAC contigs. Comparison of genetic and synteny-based physical maps indicated that ∼50% of all genetic recombination is confined to 14% of the physical length of the chromosome arm in the distal region. The 1AS physical map provides a framework for future genetic mapping projects as well as the basis for complete sequencing of chromosome arm 1AS.
Exploring the genome of the salt-marsh Spartina maritima (Poaceae, Chloridoideae) through BAC end sequence analysis.
Plant Mol Biol. 2013 Jul 23
Read more
Authors
Ferreira de Carvalho J, Chelaifa H, Boutte J, Poulain J, Couloux A, Wincker P, Bellec A, Fourment J, Bergès H, Salmon A, Ainouche M.
Plant Mol Biol. 2013 Jul 23
Abstract
Spartina species play an important ecological role on salt marshes. Spartina maritima is an Old-World species distributed along the European and North-African Atlantic coasts. This hexaploid species (2n = 6x = 60, 2C = 3,700 Mb) hybridized with different Spartina species introduced from the American coasts, which resulted in the formation of new invasive hybrids and allopolyploids. Thus, S. maritima raises evolutionary and ecological interests. However, genomic information is dramatically lacking in this genus. In an effort to develop genomic resources, we analysed 40,641 high-quality bacterial artificial chromosome-end sequences (BESs), representing 26.7 Mb of the S. maritima genome. BESs were searched for sequence homology against known databases. A fraction of 16.91 % of the BESs represents known repeats including a majority of long terminal repeat (LTR) retrotransposons (13.67 %). Non-LTR retrotransposons represent 0.75 %, DNA transposons 0.99 %, whereas small RNA, simple repeats and low-complexity sequences account for 1.38 % of the analysed BESs. In addition, 4,285 simple sequence repeats were detected. Using the coding sequence database of Sorghum bicolor, 6,809 BESs found homology accounting for 17.1 % of all BESs. Comparative genomics with related genera reveals that the microsynteny is better conserved with S. bicolor compared to other sequenced Poaceae, where 37.6 % of the paired matching BESs are correctly orientated on the chromosomes. We did not observe large macrosyntenic rearrangements using the mapping strategy employed. However, some regions appeared to have experienced rearrangements when comparing Spartina to Sorghum and to Oryza. This work represents the first overview of S. maritima genome regarding the respective coding and repetitive components. The syntenic relationships with other grass genomes examined here help clarifying evolution in Poaceae, S. maritima being a part of the poorly-known Chloridoideae sub-family.
Link
A high density physical map of chromosome 1BL supports evolutionary studies, map-based cloning and sequencing in wheat.
Genome Biol. 2013 Jun 25;14(6):R64. [Epub ahead of print]
Read more
Authors
Philippe R, Paux E, Bertin I, Sourdille P, Choulet F, Laugier C, Simkova H, Safar J, Bellec A, Vautrin S, Frenkel Z, Cattonaro F, Magni F, Scalabrin S, Martis MM, Mayer KF, Korol A, Berges H, Dolezel J, Feuillet C.
Genome Biol. 2013 Jun 25;14(6):R64. [Epub ahead of print]
Abstract
BACKGROUND:
As for other major crops, achieving a complete wheat genome sequence is essential for the application of genomics to breeding new and improved varieties. To overcome the complexities of the large, highly repetitive and hexaploid wheat genome, the International Wheat Genome Sequencing Consortium established a chromosome-based strategy that was validated by the construction of the physical map of chromosome 3B. Here, we present improved strategies for the construction of highly integrated and ordered wheat physical maps, using chromosome 1BL as a template, and illustrate their potential for evolutionary studies and map-based cloning.
RESULTS:
Using a combination of novel high throughput marker assays and an assembly program, we developed a high quality physical map representing 93% of wheat chromosome 1BL, anchored and ordered with 5,489 markers including 1,161 genes. Analysis of the gene space organization and evolution revealed that gene distribution and conservation along the chromosome results from the superimposition of the ancestral grass and recent wheat evolutionary patterns leading to a peak of synteny in the central part of the chromosome arm and an increased density of non collinear genes towards the telomere. With a density of about 11 markers per Mb, the 1BL physical map provides 916 markers, including 193 genes, for fine mapping the 40 QTLs mapped on this chromosome.
CONCLUSIONS:
Here, we demonstrate that high marker density physical maps can be developed in complex genomes such as wheat to accelerate map-based cloning, gain new insights into genome evolution, and provide a foundation for reference sequencing.:
MtQRRS1, an R-locus required for Medicago truncatula quantitative resistance to Ralstonia solanacearum.
New Phytol. 2013 May 2. doi: 10.1111/nph.12299.
Read more
Authors
Ben C, Debellé F, Berges H, Bellec A, Jardinaud MF, Anson P, Huguet T, Gentzbittel L, Vailleau F.
New Phytol. 2013 May 2. doi: 10.1111/nph.12299.
Abstract
Ralstonia solanacearum is a major soilborne pathogen that attacks > 200 plant species, including major crops. To characterize MtQRRS1, a major quantitative trait locus (QTL) for resistance towards this bacterium in the model legume Medicago truncatula, genetic and functional approaches were combined. QTL analyses together with disease scoring of heterogeneous inbred families were used to define the locus. The candidate region was studied by physical mapping using a bacterial artificial chromosome (BAC) library of the resistant line, and sequencing. In planta bacterial growth measurements, grafting experiments and gene expression analysis were performed to investigate the mechanisms by which this locus confers resistance to R. solanacearum. The MtQRRS1 locus was localized to the same position in two recombinant inbred line populations and was narrowed down to a 64 kb region. Comparison of parental line sequences revealed 15 candidate genes with sequence polymorphisms, but no evidence of differential gene expression upon infection. A role for the hypocotyl in resistance establishment was shown. These data indicate that the quantitative resistance to bacterial wilt conferred by MtQRRS1, which contains a cluster of seven R genes, is shared by different accessions and may act through intralocus interactions to promote resistance.
Physical Mapping Integrated with Syntenic Analysis to Characterize the Gene Space of the Long Arm of Wheat Chromosome 1A.
PLoS One. 2013 Apr 16;8(4):e59542. doi: 10.1371/journal.pone.0059542.
Read more
Authors
Lucas SJ, Akpınar BA, Kantar M, Weinstein Z, Aydınoğlu F, Safář J, Simková H, Frenkel Z, Korol A, Magni F, Cattonaro F, Vautrin S, Bellec A, Bergès H, Doležel J, Budak H.
PLoS One. 2013 Apr 16;8(4):e59542. doi: 10.1371/journal.pone.0059542.
Abstract
BACKGROUND:
Bread wheat (Triticum aestivum L.) is one of the most important crops worldwide and its production faces pressing challenges, the solution of which demands genome information. However, the large, highly repetitive hexaploid wheat genome has been considered intractable to standard sequencing approaches. Therefore the International Wheat Genome Sequencing Consortium (IWGSC) proposes to map and sequence the genome on a chromosome-by-chromosome basis.
METHODOLOGY PRINCIPAL FINDINGS:
We have constructed a physical map of the long arm of bread wheat chromosome 1A using chromosome-specific BAC libraries by High Information Content Fingerprinting (HICF). Two alternative methods (FPC and LTC) were used to assemble the fingerprints into a high-resolution physical map of the chromosome arm. A total of 365 molecular markers were added to the map, in addition to 1122 putative unique transcripts that were identified by microarray hybridization. The final map consists of 1180 FPC-based or 583 LTC-based contigs.
CONCLUSIONS SIGNIFICANCE:
The physical map presented here marks an important step forward in mapping of hexaploid bread wheat. The map is orders of magnitude more detailed than previously available maps of this chromosome, and the assignment of over a thousand putative expressed gene sequences to specific map locations will greatly assist future functional studies. This map will be an essential tool for future sequencing of and positional cloning within chromosome 1A.
A physical, genetic and functional sequence assembly of the barley genome.
Nature. 2012 Oct 17. doi: 10.1038/nature11543.
Read more
Authors
The International Barley Genome Sequencing Consortium; Principal investigators, Mayer KF, Waugh R, Langridge P, Close TJ, Wise RP, Graner A, Matsumoto T, Sato K, Schulman A, Muehlbauer GJ, Stein N; Physical map construction and direct anchoring, Ariyadasa R, Schulte D, Poursarebani N, Zhou R, Steuernagel B, Mascher M, Scholz U, Shi B, Langridge P, Madishetty K, Svensson JT, Bhat P, Moscou M, Resnik J, Close TJ, Muehlbauer GJ, Hedley P, Liu H, Morris J, Waugh R, Frenkel Z, Korol A, Bergès H, Graner A, Stein N; Genomic sequencing and assembly, Steuernagel B, Scholz U, Taudien S, Felder M, Groth M, Platzer M, Stein N; BAC sequencing and assembly, Steuernagel B, Scholz U, Himmelbach A, Taudien S, Felder M, Platzer M, Lonardi S, Duma D, Alpert M, Cordero F, Beccuti M, Ciardo G, Ma Y, Wanamaker S, Close TJ, Stein N; BAC-end sequencing, Cattonaro F, Vendramin V, Scalabrin S, Radovic S, Wing R, Schulte D, Steuernagel B, Morgante M, Stein N, Waugh R; Integration of physical/genetic map and sequence resources, Nussbaumer T, Gundlach H, Martis M, Ariyadasa R, Poursarebani N, Steuernagel B, Scholz U, Wise RP, Poland J, Stein N, Mayer KF; Gene annotation, Spannagl M, Pfeifer M, Gundlach H, Mayer KF; Repetitive DNA analysis, Gundlach H, Moisy C, Tanskanen J, Scalabrin S, Zuccolo A, Vendramin V, Morgante M, Mayer KF, Schulman A; Transcriptome sequencing and analysis, Pfeifer M, Spannagl M, Hedley P, Morris J, Russell J, Druka A, Marshall D, Bayer M, Swarbreck D, Sampath D, Ayling S, Febrer M, Caccamo M, Matsumoto T, Tanaka T, Sato K, Wise RP, Close TJ, Wannamaker S, Muehlbauer GJ, Stein N, Mayer KF, Waugh R; Re-sequencing and diversity analysis, Steuernagel B, Schmutzer T, Mascher M, Scholz U, Taudien S, Platzer M, Sato K, Marshall D, Bayer M, Waugh R, Stein N; Writing and editing of the manuscript, Mayer KF, Waugh R, Brown JW, Schulman A, Langridge P, Platzer M, Fincher GB, Muehlbauer GJ, Sato K, Close TJ, Wise RP, Stein N.
Nature. 2012 Oct 17. doi: 10.1038/nature11543.
Abstract
Barley (Hordeum vulgare L.) is among the world's earliest domesticated and most important crop plants. It is diploid with a large haploid genome of 5.1 gigabases (Gb). Here we present an integrated and ordered physical, genetic and functional sequence resource that describes the barley gene-space in a structured whole-genome context. We developed a physical map of 4.98 Gb, with more than 3.90 Gb anchored to a high-resolution genetic map. Projecting a deep whole-genome shotgun assembly, complementary DNA and deep RNA sequence data onto this framework supports 79,379 transcript clusters, including 26,159 'high-confidence' genes with homology support from other plant genomes. Abundant alternative splicing, premature termination codons and novel transcriptionally active regions suggest that post-transcriptional processing forms an important regulatory layer. Survey sequences from diverse accessions reveal a landscape of extensive single-nucleotide variation. Our data provide a platform for both genome-assisted research and enabling contemporary crop improvement.
Links :
http://www.ncbi.nlm.nih.gov/pubmed/23075845
http://www.international.inra.fr/press/barley_genome_sequenced
Intraspecific sequence comparisons reveal similar rates of non-collinear gene insertion in the B and D genomes of bread wheat.
BMC Plant Biol. (2012) Aug 30;12(1):155
Read more
Authors
Bartos J, Vlcek C, Choulet F, Dzunková M, Cviková K, Safár J, Simková H, Paces J, Strnad H, Sourdille P, Bergès H, Cattonaro F, Feuillet C, Dolezel J.
BMC Plant Biol. (2012) Aug 30;12(1):155
Abstract
Background:
Polyploidization is considered one of the main mechanisms of plant genome evolution. The presence of multiple copies of the same gene reduces selection pressure and permits sub-functionalization and neo-functionalization leading to plant diversification, adaptation and speciation. In bread wheat, polyploidization and the prevalence of transposable elements resulted in massive gene duplication and movement. As a result, the number of genes which are non-collinear to genomes of related species seems markedly increased in wheat.
Results:
We used new-generation sequencing (NGS) to generate sequence of a Mb-sized region from wheat chromosome arm 3DS. Sequence assembly of 24 BAC clones resulted in two scaffolds of 1,264,820 and 333,768 bases. The sequence was annotated and compared to the homoeologous region on wheat chromosome 3B and orthologous loci of Brachypodium distachyon and rice. Among 39 coding sequences in the 3DS scaffolds, 32 have a homoeolog on chromosome 3B. In contrast, only fifteen and fourteen orthologs were identified in the corresponding regions in rice and Brachypodium, respectively. Interestingly, five pseudogenes were identified among the non-collinear coding sequences at the 3B locus, while none was found at the 3DS locus.
Conclusion:
Direct comparison of two Mb-sized regions of the B and D genomes of bread wheat revealed similar rates of non-collinear gene insertion in both genomes with a majority of gene duplications occurring before their divergence. Relatively low proportion of pseudogenes was identified among non-collinear coding sequences. Our data suggest that the pseudogenes did not originate from insertion of non-functional copies, but were formed later during the evolution of hexaploid wheat. Some evidence was found for gene erosion along the B genome locus.
Down-regulation of a single auxin efflux transport protein in tomato induces precocious fruit development.
J Exp Bot. 2012 Aug;63(13):4901-17.
Read more
Authors
Mounet F, Moing A, Kowalczyk M, Rohrmann J, Petit J, Garcia V, Maucourt M, Yano K, Deborde C, Aoki K, Bergès H, Granell A, Fernie AR, Bellini C, Rothan C, Lemaire-Chamley M.
J Exp Bot. 2012 Aug;63(13):4901-17.
Abstract
The PIN-FORMED (PIN) auxin efflux transport protein family has been well characterized in the model plant Arabidopsis thaliana, where these proteins are crucial for auxin regulation of various aspects of plant development. Recent evidence indicates that PIN proteins may play a role in fruit set and early fruit development in tomato (Solanum lycopersicum), but functional analyses of PIN-silenced plants failed to corroborate this hypothesis. Here it is demonstrated that silencing specifically the tomato SlPIN4 gene, which is predominantly expressed in tomato flower bud and young developing fruit, leads to parthenocarpic fruits due to precocious fruit development before fertilization. This phenotype was associated with only slight modifications of auxin homeostasis at early stages of flower bud development and with minor alterations of ARF and Aux/IAA gene expression. However, microarray transcriptome analysis and large-scale quantitative RT-PCR profiling of transcription factors in developing flower bud and fruit highlighted differentially expressed regulatory genes, which are potential targets for auxin control of fruit set and development in tomato. In conclusion, this work provides clear evidence that the tomato PIN protein SlPIN4 plays a major role in auxin regulation of tomato fruit set, possibly by preventing precocious fruit development in the absence of pollination, and further gives new insights into the target genes involved in fruit set.
Deciphering the genomic structure, function and evolution of carotenogenesis related phytoene synthases in grasses
BMC Genomics. 2012 Jun 6;13:221.
Read more
Authors
Dibari B, Murat F, Chosson A, Gautier V, Poncet C, Lecomte P, Mercier I, Bergès H, Pont C, Blanco A, Salse J.
BMC Genomics. 2012 Jun 6;13:221.
Abstract
Background :
Carotenoids are isoprenoid pigments, essential for photosynthesis and photoprotection in plants. The enzyme phytoene synthase (PSY) plays an essential role in mediating condensation of two geranylgeranyl diphosphate molecules, the first committed step in carotenogenesis. PSY are nuclear enzymes encoded by a small gene family consisting of three paralogous genes (PSY1-3) that have been widely characterized in rice, maize and sorghum.
Results :
In wheat, for which yellow pigment content is extremely important for flour colour, only PSY1 has been extensively studied because of its association with QTLs reported for yellow pigment whereas PSY2 has been partially characterized. Here, we report the isolation of bread wheat PSY3 genes from a Renan BAC library using Brachypodium as a model genome for the Triticeae to develop Conserved Orthologous Set markers prior to gene cloning and sequencing. Wheat PSY3 homoeologous genes were sequenced and annotated, unravelling their novel structure associated with intron-loss events and consequent exonic fusions. A wheat PSY3 promoter region was also investigated for the presence of cis-acting elements involved in the response to abscisic acid (ABA), since carotenoids also play an important role as precursors of signalling molecules devoted to plant development and biotic/abiotic stress responses. Expression of wheat PSYs in leaves and roots was investigated during ABA treatment to confirm the up-regulation of PSY3 during abiotic stress.
Conclusions :
We investigated the structural and functional determinisms of PSY genes in wheat. More generally, among eudicots and monocots, the PSY gene family was found to be associated with differences in gene copy numbers, allowing us to propose an evolutionary model for the entire PSY gene family in Grasses.
Read more
Authors
Kane, N.C., Gill, N., King, M.G., Bowers, J.E., Berges, H., Gouzy, J., Bachlava, E., Langlade, N.B., Lai, Z., Stewart, M., Burke, J.M., Vincourt, P., Knapp, S.J., and Rieseberg L.H.
Botany (89:429-437)
Abstract
The Compositae is one of the largest and most economically important families of flowering plants and includes a diverse array of food crops, horticultural crops, medicinals, and noxious weeds. Despite its size and economic importance, there is no reference genome sequence for the Compositae, which impedes research and improvement efforts. We report on progress toward sequencing the 3.5 Gb genome of cultivated sunflower (Helianthus annuus), the most important crop in the family. Our sequencing strategy combines whole-genome shotgun sequencing using the Solexa and 454 platforms with the generation of high-density genetic and physical maps that serve as scaffolds for the linear assembly of whole-genome shotgun sequences. The performance of this approach is enhanced by the construction of a sequence-based physical map, which provides unique sequence-based tags every 5–6 kb across the genome. Thus far, our physical map covers ~ 85% of the sunflower genome, and we have generated ~ 80× genome coverage with Solexa reads and 15.5× with 454 reads. Preliminary analyses indicated that ~ 78% of the sunflower genome consists of repetitive sequences. Nonetheless, ~ 76% of contigs >5 kb in size can be assigned to either the physical or genetic map or to both, suggesting that our approach is likely to deliver a highly accurate and contiguous reference genome for sunflower.
The Sugarcane Genome Challenge: Strategies for Sequencing a Highly Complex Genome.
(2011) Tropical Plant Biology 4: 145-156, DOI: 10.1007/s12042-011-9079-0
Read more
Authors
Glaucia Mendes Souza, Helene Berges, Stephanie Bocs, Rosanne Casu, Angelique D’Hont, João Eduardo Ferreira, Robert Henry, Ray Ming, Bernard Potier and Marie-Anne Van Sluys, Michel Vincentz and Andrew H. Paterson
(2011) Tropical Plant Biology 4: 145-156, DOI: 10.1007/s12042-011-9079-0
Abstract
Sugarcane cultivars derive from interspecific hybrids obtained by crossing Saccharum officinarum and Saccharum spontaneum and provide feedstock used worldwide for sugar and biofuel production. The importance of sugarcane as a bioenergy feedstock has increased interest in the generation of new cultivars optimised for energy production. Cultivar improvement has relied largely on traditional breeding methods, which may be limited by the complexity of inheritance in interspecific polyploidy hybrids, and the time-consuming process of selection of plants with desired agronomic traits. In this sense, molecular genetics can assist in the process of developing improved cultivars by generating molecular markers that can be used in the breeding process or by introducing new genes into the sugarcane genome. For meeting each of these, and additional goals, biotechnologists would benefit from a reference genome sequence of a sugarcane cultivar. The sugarcane genome poses challenges that have not been addressed in any prior sequencing project, due to its highly polyploid and aneuploid genome structure with a complete set of homeologous genes predicted to range from 10 to 12 copies (alleles) and to include representatives from each of two different species. Although sugarcane’s monoploid genome is about 1 Gb, its highly polymorphic nature represents another significant challenge for obtaining a genuine assembled monoploid genome. With a rich resource of expressed-sequence tag (EST) data in the public domain, the present article describes tools and strategies that may aid in the generation of a reference genome sequence.
KeywordsSugarcane–Genome–Sequencing–Sorghum
Contrasted Patterns of Molecular Evolution in Dominant and Recessive Self-Incompatibility Haplotypes in Arabidopsis.
PLoS Genet.2012 Mar;8(3):e1002495.
PMID: 22457631
Read more
Authors
Goubet PM, Bergès H, Bellec A, Prat E, Helmstetter N, Mangenot S, Gallina S, Holl AC, Fobis-Loisy I, Vekemans X, Castric V.
PLoS Genet. 2012 Mar;8(3):e1002495.
PMID: 22457631
Abstract
Self-incompatibility has been considered by geneticists a model system for reproductive biology and balancing selection, but our understanding of the genetic basis and evolution of this molecular lock-and-key system has remained limited by the extreme level of sequence divergence among haplotypes, resulting in a lack of appropriate genomic sequences. In this study, we report and analyze the full sequence of eleven distinct haplotypes of the self-incompatibility locus (S-locus) in two closely related Arabidopsis species, obtained from individual BAC libraries. We use this extensive dataset to highlight sharply contrasted patterns of molecular evolution of each of the two genes controlling self-incompatibility themselves, as well as of the genomic region surrounding them. We find strong collinearity of the flanking regions among haplotypes on each side of the S-locus together with high levels of sequence similarity. In contrast, the S-locus region itself shows spectacularly deep gene genealogies, high variability in size and gene organization, as well as complete absence of sequence similarity in intergenic sequences and striking accumulation of transposable elements. Of particular interest, we demonstrate that dominant and recessive S-haplotypes experience sharply contrasted patterns of molecular evolution. Indeed, dominant haplotypes exhibit larger size and a much higher density of transposable elements, being matched only by that in the centromere. Overall, these properties highlight that the S-locus presents many striking similarities with other regions involved in the determination of mating-types, such as sex chromosomes in animals or in plants, or the mating-type locus in fungi and green algae.
The Medicago genome provides insight into the evolution of rhizobial symbioses.
Nature. 2011 Nov 16.
Read more
Authors
Young ND, Debellé F, Oldroyd GE, Geurts R, Cannon SB, Udvardi MK, Benedito VA, Mayer KF, Gouzy J, Schoof H, Van de Peer Y, Proost S, Cook DR, Meyers BC, Spannagl M, Cheung F, De Mita S, Krishnakumar V, Gundlach H, Zhou S, Mudge J, Bharti AK, Murray JD, Naoumkina MA, Rosen B, Silverstein KA, Tang H, Rombauts S, Zhao PX, Zhou P, Barbe V, Bardou P, Bechner M, Bellec A, Berger A, Bergès H, Bidwell S, Bisseling T, Choisne N, Couloux A, Denny R, Deshpande S, Dai X, Doyle JJ, Dudez AM, Farmer AD, Fouteau S, Franken C, Gibelin C, Gish J, Goldstein S, González AJ, Green PJ, Hallab A, Hartog M, Hua A, Humphray SJ, Jeong DH, Jing Y, Jöcker A, Kenton SM, Kim DJ, Klee K, Lai H, Lang C, Lin S, Macmil SL, Magdelenat G, Matthews L, McCorrison J, Monaghan EL, Mun JH, Najar FZ, Nicholson C, Noirot C, O'Bleness M, Paule CR, Poulain J, Prion F, Qin B, Qu C, Retzel EF, Riddle C, Sallet E, Samain S, Samson N, Sanders I, Saurat O, Scarpelli C, Schiex T, Segurens B, Severin AJ, Sherrier DJ, Shi R, Sims S, Singer SR, Sinharoy S, Sterck L, Viollet A, Wang BB, Wang K, Wang M, Wang X, Warfsmann J, Weissenbach J, White DD, White JD, Wiley GB, Wincker P, Xing Y, Yang L, Yao Z, Ying F, Zhai J, Zhou L, Zuber A, Dénarié J, Dixon RA, May GD, Schwartz DC, Rogers J, Quétier F, Town CD, Roe BA.
Nature. 2011 Nov 16.
Abstract
Legumes (Fabaceae or Leguminosae) are unique among cultivated plants for their ability to carry out endosymbiotic nitrogen fixation with rhizobial bacteria, a process that takes place in a specialized structure known as the nodule. Legumes belong to one of the two main groups of eurosids, the Fabidae, which includes most species capable of endosymbiotic nitrogen fixation. Legumes comprise several evolutionary lineages derived from a common ancestor 60 million years ago (Myr ago). Papilionoids are the largest clade, dating nearly to the origin of legumes and containing most cultivated species. Medicago truncatula is a long-established model for the study of legume biology. Here we describe the draft sequence of the M. truncatula euchromatin based on a recently completed BAC assembly supplemented with Illumina shotgun sequence, together capturing ∼94% of all M. truncatula genes. A whole-genome duplication (WGD) approximately 58 Myr ago had a major role in shaping the M. truncatula genome and thereby contributed to the evolution of endosymbiotic nitrogen fixation. Subsequent to the WGD, the M. truncatula genome experienced higher levels of rearrangement than two other sequenced legumes, Glycine max and Lotus japonicus. M. truncatula is a close relative of alfalfa (Medicago sativa), a widely cultivated crop with limited genomics tools and complex autotetraploid genetics. As such, the M. truncatula genome sequence provides significant opportunities to expand alfalfa's genomic toolbox.
A 3000-loci transcription map of chromosome 3B unravels the structural and functional features of gene islands in hexaploid wheat.
Plant Physiol. 2011 Oct 27.
Read more
Authors
Rustenholz C, Choulet F, Laugier C, Safár J, Simková H, Dolezel J, Magni F, Scalabrin S, Cattonaro F, Vautrin S, Bellec A, Bergès H, Feuillet C, Paux E.
Plant Physiol. 2011 Oct 27.
Abstract
To improve our understanding of the organization and regulation of the wheat (Triticum aestivum L.) gene space, we established the first transcription map of a wheat chromosome (3B) by hybridizing a newly developed wheat expression microarray with BAC pools from a new version of the 3B physical map as well as with cDNA probes derived from 15 RNA samples. Mapping data for almost 3000 genes showed that the gene space spans the whole chromosome 3B with a twofold increase of gene density towards the telomeres due to an increase in the number of genes in islands. Comparative analyses with rice and Brachypodium revealed that these gene islands are composed mainly of genes likely originating from interchromosomal gene duplications. Gene ontology and expression profile analyses for the 3000 genes located along the chromosome revealed that the gene islands are enriched significantly in genes sharing the same function or expression profile, thereby suggesting that genes in islands acquired shared regulation during evolution. Only a small fraction of these clusters of cofunctional and coexpressed genes was conserved with rice and Brachypodium indicating a recent origin. Finally, genes with the same expression profiles in remote islands (coregulation islands) were identified suggesting long-distance regulation of gene expression along the chromosomes in wheat.
Functional features of a single chromosome arm in wheat (1AL) determined from its structure.
Funct Integr Genomics. 2011 Sep 3.
Read more
Authors :
Lucas SJ, Simková H, Safár J, Jurman I, Cattonaro F, Vautrin S, Bellec A, Berges H, Doležel J, Budak H.
Funct Integr Genomics. 2011 Sep 3.
Abstract :
Bread wheat (Triticum aestivum L.) is one of the most important crops globally and a high priority for genetic improvement, but its large and complex genome has been seen as intractable to whole genome sequencing. Isolation of individual wheat chromosome arms has facilitated large-scale sequence analyses. However, so far there is no such survey of sequences from the A genome of wheat. Greater understanding of an A chromosome could facilitate wheat improvement and future sequencing of the entire genome. We have constructed BAC library from the long arm of T. aestivum chromosome 1A (1AL) and obtained BAC end sequences from 7,470 clones encompassing the arm. We obtained 13,445 (89.99%) useful sequences with a cumulative length of
7.57 Mb, representing 1.43% of 1AL and about 0.14% of the entire A genome. The GC content of the sequences was 44.7%, and 90% of the chromosome was estimated to comprise repeat sequences, while just over 1% encoded expressed genes. From the sequence data, we identified a large number of sites suitable for development of molecular markers
(362 SSR and 6,948 ISBP) which will have utility for mapping this chromosome and for marker assisted breeding. From 44 putative ISBP markers tested 23 (52.3%) were found to be useful. The BAC end sequence data also enabled the identification of genes and syntenic blocks specific to chromosome 1AL, suggesting regions of particular functional interest and targets for future research.
Analysis of BAC end sequences in oak, a keystone forest tree species, providing insight into the composition of its genome.
BMC Genomics. 2011 Jun 6;12(1):292
Read more
Authors :
Faivre Rampant P, Lesur I, Boussardon C, Bitton F, Martin-Magniette ML, Bodenes C, Le Provost G, Berges H, Fluch S, Kremer A, Plomion C.
BMC Genomics. 2011 Jun 6;12(1):292
Abstract :
BACKGROUND:
One of the key goals of oak genomics research is to identify genes of adaptive significance. This information may help to improve the conservation of adaptive genetic variation and the management of forests to increase their health and productivity. Deep-coverage large-insert genomic libraries are a crucial tool for attaining this objective. We report herein the construction of a BAC library for Quercus robur, its characterization and an analysis of BAC end sequences.
RESULTS:
The EcoRI library generated consisted of 92,160 clones, 7% of which had no insert. Levels of chloroplast and mitochondrial contamination were below 3% and 1%, respectively. Mean clone insert size was estimated at 135 kb. The library represents 12 haploid genome equivalents and, the likelihood of finding a particular oak sequence of interest is greater than 99%. Genome coverage was confirmed by PCR screening of the library with 60 unique genetic loci sampled from the genetic linkage map. In total, about 20,000 high-quality BAC end sequences (BESs) were generated by sequencing 15,000 clones. Roughly 5.88% of the combined BAC end sequence length corresponded to known retroelements while ab initio repeat detection methods identified 41 additional repeats. Collectively, characterized and novel repeats account for roughly 8.94% of the genome. Further analysis of the BESs revealed 1,823 putative genes suggesting at least 29,340 genes in the oak genome. BESs were aligned with the genome sequences of Arabidopsis thaliana, Vitis vinifera and Populus trichocarpa. One putative collinear microsyntenic region encoding an alcohol acyl transferase protein was observed between oak and chromosome 2 of V. vinifera.
CONCLUSIONS:
This BAC library provides a new resource for genomic studies, including SSR marker development, physical mapping, comparative genomics and genome sequencing. BES analysis provided insight into the structure of the oak genome. These sequences will be used in the assembly of a future genome sequence for oak.
Advancing Eucalyptus genomics: identification and sequencing of lignin biosynthesis genes from deep-coverage BAC libraries
BMC Genomics. 2011 Mar 4;12(1):137.
Read more
Authors :
Paiva JA, Prat E, Vautrin S, Santos MD, San-Clemente H, Brommonschenkel S, Fonseca PG, Grattapaglia D, Song X, Ammiraju JS, Kudrna D, Wing RA, Freitas AT, Berges H, Grima-Pettenati J.
BMC Genomics. 2011 Mar 4;12(1):137.
Abstract :
BACKGROUND: Eucalyptus species are among the most planted hardwoods in the world because of their rapid growth, adaptability and valuable wood properties. The development and integration of genomic resources into breeding practice will be increasingly important in the decades to come. Bacterial artificial chromosome (BAC) libraries are key genomic tools that enable positional cloning of important traits, synteny evaluation, and the development of genome framework physical maps for genetic linkage and genome sequencing.
RESULTS: We describe the construction and characterization of two deep-coverage BAC libraries EG_Ba and EG_Bb obtained from nuclear DNA fragments of E. grandis (clone BRASUZ1) digested with HindIII and BstYI, respectively. Genome coverages of 17 and 15 haploid genome equivalents were estimated for EG_Ba and EG_Bb, respectively. Both libraries contained large inserts, with average sizes ranging from 135Kb (Eg_Bb) to 157Kb (Eg_Ba), very low extra-nuclear genome contamination providing a probability of finding a single copy gene [greater than or equal to] 99.99%. Libraries were screened for the presence of several genes of interest via hybridizations to high-density BAC filters followed by PCR validation. Five selected BAC clones were sequenced and assembled using the Roche GS FLX technology providing the whole sequence of the E. grandis chloroplast genome, and complete genomic sequences of important lignin biosynthesis genes.
CONCLUSIONS: The two E. grandis BAC libraries described in this study represent an important milestone for the advancement of Eucalyptus genomics and forest tree research. These BAC resources have a highly redundant genome coverage (>15x), contain large average inserts and have a very low percentage of clones with organellar DNA or empty vectors. These publicly available BAC libraries are thus suitable for a broad range of applications in genetic and genomic research in Eucalyptus and possibly in related species of Myrtaceae, including genome sequencing, gene isolation, functional and comparative genomics. Because they have been constructed using the same tree (E. grandis BRASUZ1) whose full genome is being sequenced, they should prove instrumental for assembly and gap filling of the upcoming Eucalyptus reference genome sequence.
Construction and characterization of two BAC libraries representing a deep-coverage of the genome of chicory (Cichorium intybus L., Asteraceae).
BMC Res Notes. 2010 Aug 11;3:225.
Read more
Abstract
BACKGROUND: The Asteraceae represents an important plant family with respect to the numbers of species present in the wild and used by man. Nonetheless, genomic resources for Asteraceae species are relatively underdeveloped, hampering within species genetic studies as well as comparative genomics studies at the family level. So far, six BAC libraries have been described for the main crops of the family, i.e. lettuce and sunflower. Here we present the characterization of BAC libraries of chicory (Cichorium intybus L.) constructed from two genotypes differing in traits related to sexual and vegetative reproduction. Resolving the molecular mechanisms underlying traits controlling the reproductive system of chicory is a key determinant for hybrid development, and more generally will provide new insights into these traits, which are poorly investigated so far at the molecular level in Asteraceae.
FINDINGS: Two bacterial artificial chromosome (BAC) libraries, CinS2S2 and CinS1S4, were constructed from HindIII-digested high molecular weight DNA of the contrasting genotypes C15 and C30.01, respectively. C15 was hermaphrodite, non-embryogenic, and S2S2 for the S-locus implicated in self-incompatibility, whereas C30.01 was male sterile, embryogenic, and S1S4. The CinS2S2 and CinS1S4 libraries contain 89,088 and 81,408 clones. Mean insert sizes of the CinS2S2 and CinS1S4 clones are 90 and 120 kb, respectively, and provide together a coverage of 12.3 haploid genome equivalents. Contamination with mitochondrial and chloroplast DNA sequences was evaluated with four mitochondrial and four chloroplast specific probes, and was estimated to be 0.024% and 1.00% for the CinS2S2 library, and 0.028% and 2.35% for the CinS1S4 library. Using two single copy genes putatively implicated in somatic embryogenesis, screening of both libraries resulted in detection of 12 and 13 positive clones for each gene, in accordance with expected numbers.
CONCLUSIONS: This indicated that both BAC libraries are valuable tools for molecular studies in chicory, one goal being the positional cloning of the S-locus in this Asteraceae species.
Authors
Gonthier L, Bellec A, Blassiau C, Prat E, Helmstetter N, Rambaud C, Huss B, Hendriks T, Bergès H, Quillet MC.
BMC Res Notes. 2010 Aug 11;3:225.
Brassica orthologs from BANYULS belong to a small multigene family, which is involved in procyanidin accumulation in the seed
Planta. 2009 Sep 17.
Read more
Abstract
As part of a research programme focused on flavonoid biosynthesis in the seed coat of Brassica napus L. (oilseed rape), orthologs of the BANYULS gene that encoded anthocyanidin reductase were cloned in B. napus as well as in the related species Brassica rapa and Brassica oleracea. B. napus genome contained four functional copies of BAN, two originating from each diploid progenitor. Amino acid sequences were highly conserved between the Brassicaceae including B. napus, B. rapa, B. oleracea as well as the model plant Arabidopsis thaliana. Along the 200 bp in 5' of the ATG codon, Bna.BAN promoters (ProBna.BAN) were conserved with AtANR promoter and contained putative cis-acting elements. In addition, transgenic Arabidopsis and oilseed rape plants carrying the first 230 bp of ProBna.BAN fused to the UidA reporter gene were generated. In the two Brassicaceae backgrounds, ProBna.BAN activity was restricted to the seed coat. In B. napus seed, ProBna.BAN was activated in procyanidin-accumulating cells, namely the innermost layer of the inner integument and the micropyle-chalaza area. At the transcriptional level, the four Bna.BAN genes were expressed in the seed. Laser microdissection assays of the seed integuments showed that Bna.BAN expression was restricted to the inner integument, which was consistent with the activation profile of ProBna.BAN. Finally, Bna.BAN genes were mapped onto oilseed rape genetic maps and potential co-localisations with seed colour quantitative trait loci are discussed.
Planta. 2009 Sep 17 - Auger et al.
Authors
Bathilde Auger, Cécile Baron, Marie-Odile Lucas, Sonia Vautrin, Hélène Bergès, Boulos Chalhoub, Alain Fautrel, Michel Renard, Nathalie Nesi.
Planta. 2009 Sep 17.
Fine Mapping and Marker Development for the Crossability Gene SKr on Chromosome 5BS of Hexaploid Wheat (Triticum aestivum L.)
Genetics. 2009 Aug 3.
Read more
Abstract
Most elite wheat varieties cannot be crossed with related species thereby restricting greatly the germplasm that can be used for alien introgression in breeding programs. Inhibition to crossability is controlled genetically and a number of QTL have been identified to date, including SKr, a strong QTL affecting crossability between wheat and rye located at the distal end of chromosome 5BS. In this study, we used a recombinant SSD population originating from a cross between the poorly crossable cultivar Courtot and the crossable line MP98 to characterize the major dominant effect of SKr and map the gene at the distal end of the chromosome near the 5B homoeologous GSP locus. Colinearity with barley and rice was used to saturate the SKr region with new markers and establish orthologous relationships with a 52 kb region on rice chromosome 12. In total, five markers were mapped within a genetic interval of 0.3 cM and 400 kb of BAC contigs were established on both sides of the gene to lay the foundation for map-based cloning of SKr. Two SSR markers completely linked to SKr were used to evaluate a collection of crossable wheat progenies originating from primary triticale breeding programs. The results confirm the major effect of SKr on crossability and the usefulness of the two markers for the efficient introgression of crossability in elite wheat varieties.
Authors
Alfares W, Bouguennec A, Balfourier F, Gay G, Bergès H, Vautrin S, Sourdille P, Bernard M, Feuillet C.
Genetics. 2009 Aug 3.