

- Arnaud Bellec
- Arnaud Bellec
- 3D pool production and screening
- BAC libraires construction
- Arnaud BELLEC
- Construction and screening of BAC libraries
- Sequencing and comparative analysis
-
Dr. Maria Lucia Carneiro Vieira
Professora Titular do Departamento de Genética Escola Superior de Agricultura "Luiz de Queiroz"
Universidade de São Paulo
Caixa Postal, 83
13400-970 Piracicaba, Brasil
Phone: 55 19 3429 4442
E-mail: mlcvieir@esalq.usp.br -
Helen Alves Penha
Doutoranda em Genética e Melhoramento de Plantas
Departamento de Genética
Lab. de Biologia Celular e Molecular de Plantas
Universidade de São Paulo - USP-ESALQ
Phone: (19) 34294442
- BAC library construction
- A gene-rich fraction analysis of the Passiflora edulis genome reveals highly conserved microsyntenic regions with two related Malpighiales species.
- The chloroplast genome of Passiflora edulis (Passifloraceae) assembled from long sequence reads: structural organization and phylogenomic studies in Malpighiales.
- Begin at the beginning: A BAC-end view of the passion fruit (Passiflora) genome.
Dr. Marie-Christine Quillet et Theo Hendriks (Directeur de Thèse)
UMR USTL/INRA1281 SADV "Stress abiotiques et différenciation des végétaux cultivés",
ERT 1016, USTL,
59655 Villeneuve d'AscqDr. Catherine Feuillet
Institut National de la Recherche Agronomique-ASP
Domaine de Crouelle
234 Avenue du Brezet
63100 Clermont-Ferrand
France
www.clermont.inra.fr/umr-aspE-mail : catherine.feuillet@clermont.inra.fr
- La construction et l’ancrage des cartes physiques du blé et de l'orge pour les chromosomes 1 et 3 qui portent un grand nombre de caractères agronomiques importants (par ex. la résistance de maladie, la production et la qualité)
- L’isolement de gènes et QTLs liés à la résistance à des maladies, le rendement et les caractères de qualité pour le blé et l'orge
- L’identification et l’exploitation de nouveaux allèles pour les gènes isolés par l'utilisation de variants naturels et de populations mutées aussi bien que le germplasm sauvage
- Le développement de nouvelles variétés pour les besoins des consommateurs et des agriculteurs à travers des approches moléculaires
- Le développement de nouveaux outils bioinformatiques pour structurer, centraliser et analyser à grande échelle les données génomiques qui seront générées dans ce projet
- La mise en place de formation pour ces nouvelles approches technologiques, le partage des résultats et les transferts technologiques à l'industrie et aux collaborateurs internationaux.
- Institut National de la Recherche Agronomique - INRA - France
- Leibniz Institute of Plant Genetics and Crop Plant Research - IPK - Germany
- Institute of Experimental Botany - IEB - Czech Republic
- GSF- National Research Center for Environment and Health - GSF - Germany
- Università degli Studi di Milano - UMIL - Italy
- University of Haifa - HU - Israel
- MTT (Maa- ja elintarviketalouden tutkimuskeskus) - MTT - Finland
- Scottish Crop Research Institute - SCRI - UK
- Sabanci University - SABA - Turkey
- National Institute of Agricultural Botany - NIAB - UK
- John Innes Centre - JIC - UK
- Universität Zürich - UZH - Switzerland
- INRA Transfert - IT - France
- Biogemma - BGA - France
- Lochow-Petkus GmbH - LP - Germany
- Istituto di Genomica Applicata - IGA - Italy
- University of Bologna -UNBO - Italy
-
Dr. Patrick Vincourt
LIPM (UMR 441-2594 INRA-CNRS)
BP 52627
Chemin de Borde Rouge - Auzeville
31326 Castanet Tolosan
Email : patrick.vincourt@toulouse.inra.fr -
Laboratoire “ Interactions Plantes Microorganismes”, UMR 441-2594(INRA-CNRS)
INRA-CNRS
31326 CASTANET TOLOSAN
Directeur du laboratoire : Pascal GAMAS
Responsable scientifique : Patrick VINCOURT UMR 1095 “ Amélioration et Santé des Plantes ”
INRA – Université Blaise Pascal
63100 CLERMONT-FERRANDDirecteur du laboratoire : Gilles CHARMET
Responsable scientifique : Felicity VEAR
UMR 1097 DIA-PC “ Diversité et évolution des Plantes Cultivées ”
INRA-IRD-Montpellier Sup Agro-Univ. Montpellier II
34060 MONTPELLIERDirecteur du laboratoire : Serge HAMON
Responsable scientifique : André BERVILLE
UMR1248 Agrosystèmes et développement territorial AGIR
INRA-INPT/ENSAT
31326 CASTANET TOLOSANDirecteur du laboratoire : Michel DURU
Responsable scientifique : Philippe GRIEU
UMR1165 Unité de Recherche en Génomique Végétale URGV
INRA – CNRS
91057 EVRY CedexDirecteur du laboratoire : Michel CABOCHE
Responsable scientifique : Jean-Pierre RENOU
UR 1258 Centre National de Ressources Génomiques Végétales
CNRGV -INRA
31326 CASTANET TOLOSANDirecteur du laboratoire/ Responsable scientifique : Helène BERGES
UR INRA 1279 “ Etude du polymorphisme des génomes végétaux ”
INRA
91 030 EVRYDirecteur du laboratoire/ Responsable scientifique : Dominique BRUNEL

FINAPEA : Fine mapping and candidate genes at a major resistance QTL to Aphanomyces euteiches in pea
FINAPEA : The project will be associated to the federative PeaMUST national Investments for the Future Consortium.
Voir plus
Project coordinator:
INRA-IGEPP
Marie-Laure PILET-NAY
65 rue de Saint-Brieuc
35042 Rennes
France
Email : Marie-Laure.Pilet-Nayel@inra.fr
Project partners:
INRA- UMR Agroécologie
17 rue Sully, BP 86510
21065 DIJON Cedex
France
Email : gregoire.aubert@inra.fr
INRA-CNRGV
Hélène Berges
Email : helene.berges@inra.fr
Abstract:
The sustainable development of grain legume in cropping systems is a major challenge for providing feed and food protein and limiting nitrogen applications in crop rotations. One of the major limiting factor to the production of grain legumes, especially pea predominant in France, is the common root rot disease, due to Aphanomyces euteiches. Previous studies dissected genetics of quantitative resistance to A. euteiches in pea and identified several main QTL. The project aims to fine map and identify candidate genes underlying a major resistance QTL to A. euteiches in pea, in order to support breeding efforts for future resistant varieties and discover putative functions underlying resistance QTL which are still poorly known in plants. The project will first fine map the target resistance QTL using Near-Isogenic-Line progenies previously produced and taking advantage of extensive marker resources recently developed in pea. Then, it will identify the physical sequence and candidate genes underlying the target QTL, using BAC libraries, recent long read sequencing technologies, qRT-PCR for gene expression analysis, and taking advantage of the ongoing progress of the pea genome sequencing project. The project will be associated to the federative PeaMUST national Investments for the Future Consortium. It will gather three partners of the Plant2Pro Carnot Institute and will allow innovative research to be developed in two major thematic axis of the Institute. Results will provide original basic knowledge about candidate genes underlying quantitative disease resistance in plants. It will provide new applied knowledge and tools for breeding of pea resistant varieties, which will be a key component of an integrated control strategy against A. euteiches, in combination with other methods of plant protection.
CNRGV involvement:
Responsible: Arnaud BELLEC
The CNRGV, responsible of the task2 of the project, will construct BAC libraries of two pea genotypes one resistant and the other susceptible to Aphanomyces euteiches.
| Fund agency: This project is funding by the Plant2Pro Carnot Institute. This institute is dedicated to integrated R&D “from laboratory to field” in the area of agricultural crop production. http://www.instituts-carnot.eu/en/carnot-institute/plant2pro |
|
IBSC : The International Barley Sequencing Consortium
The objective of the International Barley Sequencing Consortium (IBSC) is to physically map and sequence the barley gene space, and an ordered physical map linked to the genetic map to accelerate crop improvement.
Voir plus
Barley ranks fourth among the cereals in worldwide production and is widely cultivated in all temperate regions from the Arctic Circle to the tropics
The barley genome size is large, with 5.3 Gb, and it’s one of the largest in cereal crops and twice the size of the human genome. Barley is a true diploid. Highly collaborative international efforts have produced a substantial body of genetic and genomic resources.
The objective of the International Barley Sequencing Consortium (IBSC) is to physically map and sequence the barley gene space, and an ordered physical map linked to the genetic map to accelerate crop improvement.
Publications related to the project
CNRGV's responsible
IWGSC : The International Wheat Genome Sequencing Consortium
Sequencing the wheat genome has long been considered an insurmountable challenge, due to the high complexity of the wheat genome. But improving average wheat yields has become a major objective with genome sequencing as its prerequisite. The International Wheat Genome Sequencing Consortium (IWGSC) was created in 2005 by a group of wheat growers, plant scientists, and public and private breeders to change this paradigm. Today, the international public-private collaborative consortium has more than 1,500 members in 60 countries. The goal of the IWGSC is to make a high quality genome sequence of the bread wheat cv. Chinese Spring publicly available, in order to serve as a foundation for the accelerated development of improved varieties and to empower all aspects of basic and applied wheat science.
Voir plus
Sequencing the wheat genome has long been considered an insurmountable challenge, due to the high complexity of the wheat genome. But improving average wheat yields has become a major objective with genome sequencing as its prerequisite.
The International Wheat Genome Sequencing Consortium (IWGSC) was created in 2005 by a group of wheat growers, plant scientists, and public and private breeders to change this paradigm.
Today, the international public-private collaborative consortium has more than 1,500 members in 60 countries. The goal of the IWGSC is to make a high quality genome sequence of the bread wheat cv. Chinese Spring publicly available, in order to serve as a foundation for the accelerated development of improved varieties and to empower all aspects of basic and applied wheat science.
The IWGSC is a 501(c)(3) nonprofit organization registered in the United States and is led by a Board of Directors, a Leadership Team, and a Coordinating Committee. The Board of Directors decides the overall strategy and the Leadership Team is in charge of the daily management. The Coordinating Committee, composed of sponsors and leaders of IWGSC projects, is responsible for establishing the overall scientific strategy and the strategic roadmap.
The vision of the IWGSC is to establish a high quality reference sequence of the wheat genome anchored to the genetic/phenotypic maps. This will provide high resolution links between wheat traits and variations and the associated sequence features (i.e., genes, regulatory motifs, intergenic regions etc) and polymorphisms (Single Nucleotide Variants (SNPs), Structural Variations (SV)).
The IWGSC strategic roadmap has four key milestones:
1\ use of survey sequences of the 21 bread wheat chromosomes to assign gene sequences to individual chromosomes;
2\ develop physical maps to provide resources for sequencing;
3\ deliver a reference sequence for each of the chromosomes
4\ produce a gold standard genome sequence by integrating chromosome based genomic resources with the IWGSC whole genome assembly.
The first milestone was reached on July 2014 with the publication of the chromosome-based draft genome sequence in the journal Science. The physical maps for all chromosomes (milestone 2) were completed by the end of 2015. In June 2016, a whole genome shotgun assembly (IWGSC WGA v0.4) was made available pre-publication. The whole genome assembly was subsequently integrated with physical maps and other chromosome-based sequence resources to generate the first version of the chromosome-based reference sequence (RefSeq v1.0), which was made available pre-publication in January 2017 (milestone 3 & 4).
Work is now focused on delivering a high quality reference genome sequence that is anchored to the genetic maps, integrates different data resources, provides automated and manual annotation of genes and genomic features, and links genomic data directly to agronomically important traits.
IWGSC links:
Home page: http://www.wheatgenome.org/
News page: http://www.wheatgenome.org/News/Latest-news
Project section: http://www.wheatgenome.org/Projects
Facebook: https://www.facebook.com/wheat.genome
Twitter: https://twitter.com/wheatgenome
Why sequencing the wheat genome is crucial?
http://newfoodeconomy.com/cracking-the-wheat-genome/
CNRGV's responsible
Arnaud Bellec
Publications related to the project :

SUNRISE : SUNflower Resources to Improve yield Stability in a changing Environment
Project funded in the frame of "Investments for the future"
Voir plus
The world oilseed production will be faced to an increasing demand in the next thirty years catapulted by a combination of factors, including: i) an additional demand of edible oil, ii) the development of the biofuels industry and more specifically biodiesel around the world, and iii) the needs for green chemistry.
Sunflower represents a major renewable resource for food (oil), feed (meal), and green energy. France is a major sunflower producer. French breeders are ranked first in terms of sunflower seed production. World leading seed companies and SME involved in sunflower breeding are based in France and are creating a wide range of employments in France – from molecular breeders to farmers involved in seed production - , while ensuring a continuous genetic progress in integrating new technologies in their breeding programs.
During the past ten years, the impact of genetic advance on sunflower yield increase has been lower than expected, suggesting that current breeding resources and methods might not bring future solutions to the requested need in a context of climate changes. In order to face the challenges of delivering safe and high-quality food while maintaining yield and stability across different environments affected by climatic change, a paradigm shift is needed in sunflower breeding.
The near availability of the genome sequences of Helianthus annuus together with the breakthrough created by the new sequencing technologies, the development of phenotypic platforms and of bioinformatic tools allowing the integration of such high throughput data, are offering a favorable context to reinforce, through an optimization of the breeding process of hybrid cultivars, the competitiveness of French seed industry.
In an unprecedented effort (8 years project, 10 public and 7 private partners), SUNRISE offers unique opportunities to accelerate genetic gain and improve oil yield of sunflower hybrids grown under limited water supply through a better use of the outstanding genetic diversity of wild and domesticated Helianthus annuus genetic resources.
As the result of climate changes, more variability is expected in the timing and quantity of water availability for crop production from location to location. Sunflower is seen as a water stress tolerant crop, but wastes available water. On the long term period, due to the cost of investments all along the oil industry chain, the competitiveness of the sunflower chain is highly depending on the stability of oil yield across years and locations. SUNRISE will decipher the genes and genes networks involved in both the sunflower oil yield potential and its stability across years and locations, and then identify in the wide Helianthus annuus gene pool which alleles would be of interest for sunflower breeding.
Moreover, SUNRISE will devote a particular effort in the improvement of sunflower hybrid breeding process. SUNRISE will identify the loci and/or the heterosis mechanisms which are the most involved in homeostasis, depending on the parental alleles, and then build new gene pools exhibiting between themselves the better specific combining ability for homeostasis, i.e. yield stability.
All together, these two objectives will result in the definition of a new sunflower ideotype.
Full benefit of present possibilities requires (i) the better characterization of genetic diversity in order to optimize dense genotyping needed for genome wide association and linkage mapping, (ii) the development of appropriate detailed or high throughput phenotyping strategies, including molecular phenotyping, to characterize the sunflower response to the variation of the abiotic environment, (iii) the involvement of appropriate genetic design to decipher the genetic factors involved in this response, (iv) the integration of the knowledge into a crop model for in silico testing of the genotype environment interactions and for ideotypes design, (v) the development of innovative tools and methods to optimize the utilization of genetic resources and breed for new improved sunflower varieties for the concerned traits.
It relies on a large partnership between the key players of sunflower economy in France and in particular a strong private partnership unique to date in the history of sunflower research in France. This partnership will ensure that the new knowledge, resources and methods resulting from the project will be translated into products and varieties supporting the sunflower economy in France.
SUNRISE will ultimately enable efficient molecular based improvement to reduce the time from trait to commercialization. Indeed, SUNRISE will not only produce new resources, data and material, but will also follow their valorization by partners (usually competitors) that agree to synergize their efforts within the partnership of this project and give a feedback of their value in their programs. The project also integrates socio-economic analyses that will enable to evaluate the benefit of innovative genetic and ecophysiological results for the breeding sector and transfer of knowledge to agriculture.
Project coordinator:
Patrick VINCOURT
INRA UMR INRA-CNRS 441-2559
Chemin de Borde Rouge
31326 Castanet Tolosan
Patrick.Vincourt@toulouse.inra.fr
Project partner:
10 public and 7 private partners (CETIOM, BIOGEMMA,CAUSSADE SEMENCES, MAISADOUR, RAGT, SOLTIS,SYNGENTA SEEDS)
CNRGV involvement:
Responsible:
Arnaud Bellec
William Marande
Sonia Vautrin
Production and analysis of new genomic resources
![]()

Breedwheat : Developing new wheat varieties for sustainable agriculture
Developing new wheat varieties for sustainable agriculture.
Voir plus
The project aims at strengthening the competitiveness of the French breeding sector while addressing the societal demand for sustainability, quality and safety. Bringing together 26 partners from the public and private research and breeding sectors, it has a total budget of €34 million for 9 years.
Led by Catherine Feuillet, Research Director at INRA Clermont-Theix, Breedwheat will combine structural and functional genomics, genetics, and ecophysiology with high throughput phenotyping and genotyping to identify markers and genes underlying yield and quality traits under abiotic and biotic stress. Moreover, the project will characterize and tap unexploited genetic resources to expand the diversity of French gene banks. Finally, new breeding methods will be developed and evaluated for their socio-economic impact. .This project will enhance the sustainability of wheat production through the development of new varieties that are more robust and less demanding in water and fertilizer, towards a more environmentally friendly agriculture that is adapted to climate change.
Breedwheat will reinforce the French position in the “Wheat-Global Alliance” for world food security led by the international research centers CIMMYT and ICARDA. It will also enable new collaborations with other large scale national projects in the framework of the new international “Wheat Initiative” for the coordination of wheat research, supported by the G20, co-led by INRA and launched on September 15th, 2011 at the French Ministry of Agriculture.
Breedwheat is the 1st French Stimulus Initiative project coordinated by INRA in the category “Biotechnologies and Bioresources” to be launched. Announced on February 23rd, 2011 by the French Minister of Higher Education and Research and the French Minister of Agriculture, Breedwheat is one of the five laureates of the “Biotechnologies and Bioresources” call for proposals. The project represents a total investment of €34 million for 9 years, of which €9 million were granted by the French Stimulus Initiative through the French National Research Agency (ANR). The project gathers the competencies of 26 French partners, including 11 private companies, to develop and use efficient genome sequence-based tools and new methodologies for breeding wheat varieties with improved quality, sustainability, and productivity.
Project coordinator:
INRA - UMR 1095 Génétique, Diversité, Ecophysiologie des Céréales (GDEC)
5, chemin de Beaulieu
63100 Clermont-Ferrand
France
![]()
Project partners:
The project involves 26 French partners, including 11 private companies.
CNRGV's responsible
CNRGV involvement:
PAGE : Plant and Animal Genome Evolution
Plant and Animal Genome Evolution (PAGE)
Voir plus
Project coordinator:
INRA-GDEC
Jérome SALSE
Site du Crouël
234 avenue du Brézet
63100 Clermont Ferrand
Project partners:
UR1164 URGI Unité de Recherche Génomique-Info
Hadi Quesneville
route de Saint-Cyr
78026 Versailles
INRA-LGC
Thomas Faraut
Chemin de Borde Rouge
31326 Castatnet Tolosan
CNRS-LGDP
Richard Cooke
Laboratoire Genome et Developpement des Plantes
58, Avenue P. Alduy
66860 Perpignan
DYOGEN group
CNRS UMR8541
Hugues Roest Crollius
Ecole Normale Supérieure
46 rue d’Ulm
75005 Paris
INRA-CNRGV
Hélène Berges
Chemin de Borde Rouge
31326 Castanet Tolosan
Abstract:
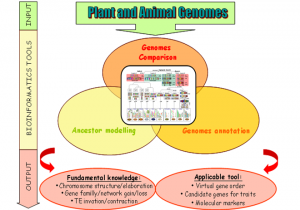
Given the socio-economic impact of plant and animals and the recent explosion of worldwide sequencing capacity, it is essential to develop tools to handle what will become a massive amount of information from sequenced genomes.The current proposal aims at developing tools to compare genomes to gain insight into plant and animal genome evolution since their common ancestors in terms of genome organisation (gene and repeated sequences) and the development of applicable tools for translational genomics purposes, i.e. transferring information (candidate genes, molecular markers) from sequenced genomes to non-sequenced ones, especially for genetic/physical mapping and future genome sequencing. The PAGE program will develop resources and approaches in comparative genomics and deliver public applicable tools for the scientific community.
The program involves 6 French academic partners from INRA (4 research groups) and CNRS (2 research groups) recognized through their publication records and ongoing collaborations as pioneers and major scientific groups in the field of plant and animal evolutionary genomics.
CNRGV's responsible
CNRGV involvement
Publication related to the project

Passiflora BAC library construction
Thesis on "Genetic-chromosomic maps of Passiflora"
Voir plus
In collaboration with:
Website : http://www.esalq.usp.br/
Abstract
Passiflorae ( Passifloraceae) is a large and widespread genus consisting primarily of tropical flora. Many Passiflora are cultivated as ornamentals and for their edible fruits or medicinal properties. The demand of aromatic passion fruit juice in the European and American market is very encouraging to South American producers. Then, the selection of varieties that combine high productivity, fruit quality and resistance to diseases, mainly to the leaf spot caused by Xanthomonas axonopodis is of great interest. Our group at University of Sao Paulo (ESALQ, Brazil) has been working on the construction of Passiflora edulis linkage maps at well as bacterial resistance was assessed in the mapping population, leading to the detection of at least one significant quantitative resistance locus. However, the development of genetic maps for auto-incompatible species, such as the passion fruit is restricted due to the unfeasibility of obtaining traditional mapping populations based on inbred lines. For this reason, the objective of our study is to construct a new integrated molecular map combining different loci configuration, using codominant markers and a novel approach based on simultaneous maximum-likehood estimation of linkage and linkage phases, specially designed for outcrossing species. Moreover, we provided the identification of P. edulis chromosomes (n= 9) based on conventional staining and in situ hybridization of repetitive DNA sequences. Then, we plan to obtain BAC libraries to refine our work to search for microsatellite and SNP markers, to sequencing, identification and mapping of resistance genes, and to allocate linkage groups to the chromosomes of the species; by searching for the linked markers in the BACs we will be able to use them as FISH probes.
Responsible : Arnaud Bellec / Nicolas Helmstetter
CNRGV involvement
Publication related to the project:

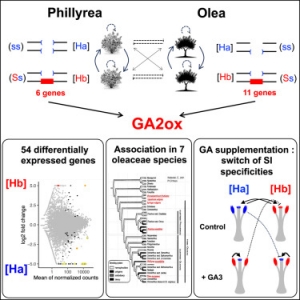
Cartographie génétique et physique du locus S chez la chicorée (Asteraceae)
Thèse en collaboration avec l'université de Lille.
Voir plus
En collaboration avec
Site web : http://www.univ-lille1.fr/pdv/labo/
Résumé
L'identification des facteurs génétiques et des mécanismes qui déterminent les étapes clés de la reproduction des plantes supérieures est l'un des thèmes nouvellement validé de l'UMR 1281 INRA/USTL 'Stress Abiotiques et Différenciation des Végétaux cultivés'.
De nombreuses espèces végétales hermaphrodites ont développé des mécanismes d’auto-incompabibilité (AI), afin de limiter les effets délétères de la consanguinité. L’incompatibilité est souvent déterminée par un locus majeur complexe, multi-allélique, le locus S. Les déterminants polliniques et stigmatiques de la réaction d’incompatibilité ont été identifiés dans quelques familles botaniques et sont de nature différentes. Ces résultats suggèrent fortement que différents mécanismes moléculaires de l’AI sont apparus de façon indépendante au cours de l'évolution des plantes à fleurs. La connaissance de cette variabilité est importante pour comprendre les bases moléculaires des interactions cellule à cellule chez les plantes.
La chicorée ( Cichorium intybus L.) appartient à la famille des Asteraceae. L’AI est très répandue dans cette espèce, non seulement chez les formes spontanées mais aussi chez les formes cultivées (chicorées endives, chicorée cultivées pour la valorisation de leurs racines appelées chicorée industrielles). Quelques rares études empiriques, indiquent que l’AI de la chicorée est déterminée par un locus majeur, le locus S. Cependant, les déterminants de l'incompatibilité sont encore inconnus. Outre l’intérêt fondamental d’identifier les gènes impliqués chez les Asteraceae, l’étude de ce caractère chez la chicorée présente un intérêt pour la création variétale, active dans notre région. L’enjeu est de disposer, pour les sélectionneurs, d’outils permettant de crééer des lignées à forte autofertilité qui seront sélectionnées en vue de la création de variétés hybrides stables et performantes.
A partir d’une descendance intra-spécifique de chicorées industrielles (K28´K59) , nous avons déterminé le profil de compatibilité d'environ 300 individus en réalisant des croisements contrôlés avec des plantes testeurs. Parallèlement, une carte génétique de la descendance K28´K59 est réalisée. La carte génétique comprend actuellement plus de 400 marqueurs, répartis sur les 9 chromosomes du génome haploïde de la chicorée. Le locus S a été cartographié à l'extrémité de l'un de ces 9 groupes de liaison dans un intervalle de 3 cM (centi Morgan).
Les objectifs de la thèse sont de réaliser une une carte génétique de haute densité autour du locus S et de réaliser une carte physique de la région du locus S, s'appuyant sur les données de cartographie génétique et la disponibilité d'une banque d'ADN génomique de type BAC ( Bacterial Artificial Chromosome). A terme, ces travaux permettront de réaliser le clonage positionnel des déterminants polliniques et stigmatiques de l'AI chez la chicorée.
Techniques mis en oeuvre par le CNRGV
Responsable : Arnaud Bellec
- Construction banque BAC
- Production Macroarray
TriticaeGenome : Vers la caractérisation du génome du blé et de l'orge
Vers la caractérisation du génome du blé et de l'orge
Voir plus
![]()
Seventh framework programmeFood Agriculture and Fisheries, Biotechnology
Coordinateur du projet
Résumé
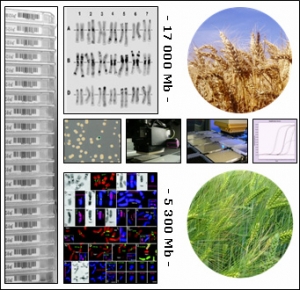
Depuis de nombreuses années, la grandeur et la complexité des génomes du blé, de l'orge et du seigle ont ralenti le développement des approches génomiques et de leurs applications dans l’amélioration de la production et la qualité des récoltes. Cependant, récemment, de nouvelles approches technologiques plus efficaces ont été développées et de nouvelles ressources on été générées qui permettent que des programmes de génomiques robustes puissent être envisagés pour les Triticeae.
TriticeaeGenome est conçu pour accomplir des progrès significatifs dans la génomique des Triticeae et soutenir la reproduction efficace de variétés améliorées pour l'agriculture européenne. Ce projet sera développé par :
Triticeaegenome est mis en place comme une contribution majeure aux efforts de consortiums internationaux dans le fait de construire des cartes physiques d'orge et de blé hexaploïde pour l’amélioration de l’agriculture, l'accélération de l'isolement de gènes et QTL et la mise en place de bases solides pour les prochains programmes de séquençage de ces génomes.
Triticeaegenome donnera des informations et des outils originaux aux éleveurs et aux scientifiques pour une meilleure compréhension de l'organisation des génomes des Triticeae, leur évolution et leur fonction, en fournissant une meilleure compréhension de la biologie de ces plantes cultivées essentielles.
liens utiles :
http://tritigen.ari.gov.cy/
http://www.wheatgenome.org/
http://urgi.versailles.inra.fr/Projects/TriticeaeGenome
Implications du CNRGV
Responsable : Arnaud Bellec/ Sonia Vautrin
- Conservation de banques BAC chromosome spécifique du blé.
- Rearrangement du Minimum Tilling Path
- Production de pools 3D sur les banques BACs chromosomes spécifiques Blé et sur la banque BAC Orge
Laboratoires partenaires

SUNYFUEL : Améliorer la qualité et les rendements de production du tournesol pour les biocarburants par des approches concertées de génétique et génomique
Améliorer la qualité et les rendements de production du tournesol pour les biocarburants par des approches concertées de génétique et génomique
Voir plus
![]()
![]()
Projet ANR- Réseau de Génomique Végétale - Génoplante 2010
Coordinateur du projet
Résumé :
Du fait de ses caractéristiques agronomiques et technologiques, le tournesol “ oléique ” est appelé à prendre dans la sole européenne une place de choix parmi les cultures respectueuses de l‘environnement dédiées à la production de matière première pour les biocarburants de première génération, sous réserve que soient levés les facteurs génétiques qui limitent son potentiel de production dans un système de culture à bas intrants. Ce projet vise à valoriser conjointement les outils de génomique créés par la communauté internationale et les ressources génétiques de type cultivé ou sauvage du genre Helianthus, pour identifier le polymorphisme exploitable de gènes clefs pour la tolérance aux stress abiotiques, cible privilégiée pour l’amélioration de la productivité en conditions contraintes, et pour l’optimisation du profil qualitatif de la récolte. Pour atteindre cet objectif seront développées les démarches de génétique d’association à partir de différentes core collections, de recherche de nouveaux gènes candidats par l’analyse et la modélisation des interactions entre niveaux d’expression et caractéristiques de l’environnement abiotique, de cartographie fine de QTL déjà mis en évidence, et de validation puis d’inventaire du polymorphisme de facteurs modificateurs de la teneur en acide oléique de la graine.
Liens :
http://lipm-helianthus.toulouse.inra.fr/dokuwiki/doku.php?id=start
http://www.heliagene.org/
http://www.heliagene.org/cgi/heliagene.cgi?__wb_session=WB12046189474944&__wb_main_menu=Collections&__wb_function=Collections
Implications du CNRGV :
Responsable: Arnaud BELLEC
- Construction d'une banque BAC tournesol
Laboratoires Partenaires :
- Frontiers in Plant Science




Capturing Wheat Phenotypes at the Genome Level
Babar Hussain, Bala A. Akpınar, Michael Alaux, Ahmed M. Algharib, Deepmala Sehgal, Zulfiqar Ali, Gudbjorg I. Aradottir, Jacqueline Batley, Arnaud Bellec, Alison R. Bentley, Halise B. Cagirici, Luigi Cattivelli, Fred Choulet, James Cockram, Francesca Desiderio, Pierre Devaux, Munevver Dogramaci, Gabriel Dorado, Susanne Dreisigacker, David Edwards, Khaoula El-Hassouni, Kellye Eversole, Tzion Fahima, Melania Figueroa, Sergio Gálvez, Kulvinder S. Gill, Liubov Govta, Alvina Gul, Goetz Hensel, Pilar Hernandez, Leonardo Abdiel Crespo-Herrera, Amir Ibrahim, Benjamin Kilian, Viktor Korzun, Tamar Krugman, Yinghui Li, Shuyu Liu, Amer F. Mahmoud, Alexey Morgounov, Tugdem Muslu, Faiza Naseer, Frank Ordon, Etienne Paux, Dragan Perovic, Gadi V. P. Reddy, Jochen Christoph Reif, Matthew Reynolds, Rajib Roychowdhury, Jackie Rudd, Taner Z. Sen, Sivakumar Sukumaran, Bahar Sogutmaz Ozdemir, Vijay Kumar Tiwari, Naimat Ullah, Turgay Unver, Selami Yazar, Rudi Appels and Hikmet Budak
Voir plus
Recent technological advances in next-generation sequencing (NGS) technologies have dramatically reduced the cost of DNA sequencing, allowing species with large and complex genomes to be sequenced. Although bread wheat (Triticum aestivum L.) is one of the world’s most important food crops, efficient exploitation of molecular marker-assisted breeding approaches has lagged behind that achieved in other crop species, due to its large polyploid genome. However, an international public–private effort spanning 9 years reported over 65% draft genome of bread wheat in 2014, and finally, after more than a decade culminated in the release of a gold-standard, fully annotated reference wheat-genome assembly in 2018. Shortly thereafter, in 2020, the genome of assemblies of additional 15 global wheat accessions was released. As a result, wheat has now entered into the pan-genomic era, where basic resources can be efficiently exploited. Wheat genotyping with a few hundred markers has been replaced by genotyping arrays, capable of characterizing hundreds of wheat lines, using thousands of markers, providing fast, relatively inexpensive, and reliable data for exploitation in wheat breeding. These advances have opened up new opportunities for marker-assisted selection (MAS) and genomic selection (GS) in wheat. Herein, we review the advances and perspectives in wheat genetics and genomics, with a focus on key traits, including grain yield, yield-related traits, end-use quality, and resistance to biotic and abiotic stresses. We also focus on reported candidate genes cloned and linked to traits of interest. Furthermore, we report on the improvement in the aforementioned quantitative traits, through the use of (i) clustered regularly interspaced short-palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9)-mediated gene-editing and (ii) positional cloning methods, and of genomic selection. Finally, we examine the utilization of genomics for the next-generation wheat breeding, providing a practical example of using in silico bioinformatics tools that are based on the wheat reference-genome sequence.




The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution
Nature 546 : 148–152 doi:10.1038/nature22380
Voir plus
Hélène Badouin, Jérôme Gouzy, Christopher J. Grassa, Florent Murat, S. Evan Staton, Ludovic Cottret, Christine Lelandais-Brière, Gregory L. Owens, Sébastien Carrère, Baptiste Mayjonade, Ludovic Legrand, Navdeep Gill, Nolan C. Kane, John E. Bowers, Sariel Hubner, Arnaud Bellec, Aurélie Bérard, Hélène Bergès, Nicolas Blanchet, Marie-Claude Boniface, Dominique Brunel, Olivier Catrice, Nadia Chaidir, Clotilde Claudel, Cécile Donnadieu, Thomas Faraut, Ghislain Fievet, Nicolas Helmstetter, Matthew King, Steven J. Knapp, Zhao Lai, Marie-Christine Le Paslier, Yannick Lippi, Lolita Lorenzon, Jennifer R. Mandel, Gwenola Marage, Gwenaëlle Marchand, Elodie Marquand, Emmanuelle Bret-Mestries, Evan Morien, Savithri Nambeesan, Thuy Nguyen, Prune Pegot-Espagnet, Nicolas Pouilly, Frances Raftis, Erika Sallet, Thomas Schiex, Justine Thomas, Céline Vandecasteele, Didier Varès, Felicity Vear, Sonia Vautrin, Martin Crespi, Brigitte Mangin, John M. Burke, Jérôme Salse, Stéphane Muños, Patrick Vincourt, Loren H. Rieseberg & Nicolas B. Langlade
Abstract
The domesticated sunflower, Helianthus annuus L., is a global oil crop that has promise for climate change adaptation, because it can maintain stable yields across a wide variety of environmental conditions, including drought. Even greater resilience is achievable through the mining of resistance alleles from compatible wild sunflower relatives, including numerous extremophile species. Here we report a high-quality reference for the sunflower genome (3.6 gigabases), together with extensive transcriptomic data from vegetative and floral organs. The genome mostly consists of highly similar, related sequences and required single-molecule real-time sequencing technologies for successful assembly. Genome analyses enabled the reconstruction of the evolutionary history of the Asterids, further establishing the existence of a whole-genome triplication at the base of the Asterids II clade and a sunflower-specific whole-genome duplication around 29 million years ago. An integrative approach combining quantitative genetics, expression and diversity data permitted development of comprehensive gene networks for two major breeding traits, flowering time and oil metabolism, and revealed new candidate genes in these networks. We found that the genomic architecture of flowering time has been shaped by the most recent whole-genome duplication, which suggests that ancient paralogues can remain in the same regulatory networks for dozens of millions of years. This genome represents a cornerstone for future research programs aiming to exploit genetic diversity to improve biotic and abiotic stress resistance and oil production, while also considering agricultural constraints and human nutritional needs.
The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution
https://www.nature.com/nature/journal/v546/n7656/full/nature22380.html
Long Read Sequencing Technology to Solve Complex Genomic Regions Assembly in Plants.
Journal of Next Generation Sequencing & Applications.
Voir plus
Authors :
Arnaud Bellec, Audrey Courtial, Stephane Cauet, Nathalie Rodde, Sonia Vautrin, Genseric Beydon, Nadege Arnal, Nadine Gautier, Joelle Fourment, Elisa Prat, William Marande, Yves Barriere and Helene Berges.
Journal of Next Generation Sequencing & Applications.
Abstract :
Background:
Numerous completed or on-going whole genome sequencing projects have highlighted the fact that obtaining a high quality genome sequence is necessary to address comparative genomics questions such as structural variations among genotypes and gain or loss of specific function. Despite the spectacular progress that has been made in sequencing technologies, obtaining accurate and reliable data is still a challenge, both at the whole genome scale and when targeting specific genomic regions. These problems are even more noticeable for complex plant genomes. Most plant genomes are known to be particularly challenging due to their size, high density of repetitive elements and various levels of ploidy. To overcome these problems, we have developed a strategy to reduce genome complexity by using the large insert BAC libraries combined with next generation sequencing technologies.
Results:
We compared two different technologies (Roche-454 and Pacific Biosciences PacBio RS II) to sequence pools of BAC clones in order to obtain the best quality sequence. We targeted nine BAC clones from different species (maize, wheat, strawberry, barley, sugarcane and sunflower) known to be complex in terms of sequence assembly. We sequenced the pools of the nine BAC clones with both technologies. We compared assembly results and highlighted differences due to the sequencing technologies used.
Conclusions:
We demonstrated that the long reads obtained with the PacBio RS II technology serve to obtain a better and more reliable assembly, notably by preventing errors due to duplicated or repetitive sequences in the same region.
Link :
Begin at the beginning: A BAC-end view of the passion fruit (Passiflora) genome.
BMC Genomics. 2014 Sep 26;15:816. doi: 10.1186/1471-2164-15-816
Voir plus
Authors
Santos AA, Penha HA, Bellec A, Munhoz Cde F, Pedrosa-Harand A, Bergès H, Vieira ML.
BMC Genomics. 2014 Sep 26;15:816. doi: 10.1186/1471-2164-15-816
Abstract
Background:
The passion fruit (Passiflora edulis) is a tropical crop of economic importance both for juice production and consumption as fresh fruit. The juice is also used in concentrate blends that are consumed worldwide. However, very little is known about the genome of the species. Therefore, improving our understanding of passion fruit genomics is essential and to some degree a pre-requisite if its genetic resources are to be used more efficiently. In this study, we have constructed a large-insert BAC library and provided the first view on the structure and content of the passion fruit genome, using BAC-end sequence (BES) data as a major resource.
Results:
The library consisted of 82,944 clones and its levels of organellar DNA were very low. The library represents six haploid genome equivalents, and the average insert size was 108 kb. To check its utility for gene isolation, successful macroarray screening experiments were carried out with probes complementary to eight Passiflora gene sequences available in public databases. BACs harbouring those genes were used in fluorescent in situ hybridizations and unique signals were detected for four BACs in three chromosomes (n = 9). Then, we explored 10,000 BES and we identified reads likely to contain repetitive mobile elements (19.6% of all BES), simple sequence repeats and putative proteins, and to estimate the GC content (~42%) of the reads. Around 9.6% of all BES were found to have high levels of similarity to plant genes and ontological terms were assigned to more than half of the sequences analysed (940). The vast majority of the top-hits made by our sequences were to Populus trichocarpa (24.8% of the total occurrences), Theobroma cacao (21.6%), Ricinus communis (14.3%), Vitis vinifera (6.5%) and Prunus persica (3.8%).
Conclusions:
We generated the first large-insert library for a member of Passifloraceae. This BAC library provides a new resource for genetic and genomic studies, as well as it represents a valuable tool for future whole genome study. Remarkably, a number of BAC-end pair sequences could be mapped to intervals of the sequenced Arabidopsis thaliana, V. vinifera and P. trichocarpa chromosomes, and putative collinear microsyntenic regions were identified.
Structural and functional partitioning of bread wheat chromosome 3B.
Science. 2014 Jul 18;345(6194):1249721. doi: 10.1126/science.1249721.
Voir plus
Authors
Choulet F, Alberti A, Theil S, Glover N, Barbe V, Daron J, Pingault L, Sourdille P, Couloux A, Paux E, Leroy P, Mangenot S, Guilhot N, Le Gouis J, Balfourier F, Alaux M, Jamilloux V, Poulain J, Durand C, Bellec A, Gaspin C, Safar J, Dolezel J, Rogers J, Vandepoele K, Aury JM, Mayer K, Berges H, Quesneville H, Wincker P, Feuillet C.
Science. 2014 Jul 18;345(6194):1249721. doi: 10.1126/science.1249721.
Abstract
We produced a reference sequence of the 1-gigabase chromosome 3B of hexaploid bread wheat. By sequencing 8452 bacterial artificial chromosomes in pools, we assembled a sequence of 774 megabases carrying 5326 protein-coding genes, 1938 pseudogenes, and 85% of transposable elements. The distribution of structural and functional features along the chromosome revealed partitioning correlated with meiotic recombination. Comparative analyses indicated high wheat-specific inter- and intrachromosomal gene duplication activities that are potential sources of variability for adaption. In addition to providing a better understanding of the organization, function, and evolution of a large and polyploid genome, the availability of a high-quality sequence anchored to genetic maps will accelerate the identification of genes underlying important agronomic traits.
Sequence-Based Analysis of Structural Organization and Composition of the Cultivated Sunflower (Helianthus annuus L.) Genome.
Biology (Basel). 3(2):295-319. doi: 10.3390/biology3020295
Voir plus
Authors
Gill N, Buti M, Kane N, Bellec A, Helmstetter N, Berges H, Rieseberg LH.
Biology (Basel). 3(2):295-319. doi: 10.3390/biology3020295
Abstract
Sunflower is an important oilseed crop, as well as a model system for evolutionary studies, but its 3.6 gigabase genome has proven difficult to assemble, in part because of the high repeat content of its genome. Here we report on the sequencing, assembly, and analyses of 96 randomly chosen BACs from sunflower to provide additional information on the repeat content of the sunflower genome, assess how repetitive elements in the sunflower genome are organized relative to genes, and compare the genomic distribution of these repeats to that found in other food crops and model species. We also examine the expression of transposable element-related transcripts in EST databases for sunflower to determine the representation of repeats in the transcriptome and to measure their transcriptional activity. Our data confirm previous reports in suggesting that the sunflower genome is >78% repetitive. Sunflower repeats share very little similarity to other plant repeats such as those of Arabidopsis, rice, maize and wheat; overall 28% of repeats are "novel" to sunflower. The repetitive sequences appear to be randomly distributed within the sequenced BACs. Assuming the 96 BACs are representative of the genome as a whole, then approximately 5.2% of the sunflower genome comprises non TE-related genic sequence, with an average gene density of 18kbp/gene. Expression levels of these transposable elements indicate tissue specificity and differential expression in vegetative and reproductive tissues, suggesting that expressed TEs might contribute to sunflower development. The assembled BACs will also be useful for assessing the quality of several different draft assemblies of the sunflower genome and for annotating the reference sequence.
Exploring the genome of the salt-marsh Spartina maritima (Poaceae, Chloridoideae) through BAC end sequence analysis.
Plant Mol Biol. 2013 Jul 23
Voir plus
Authors
Ferreira de Carvalho J, Chelaifa H, Boutte J, Poulain J, Couloux A, Wincker P, Bellec A, Fourment J, Bergès H, Salmon A, Ainouche M.
Plant Mol Biol. 2013 Jul 23
Abstract
Spartina species play an important ecological role on salt marshes. Spartina maritima is an Old-World species distributed along the European and North-African Atlantic coasts. This hexaploid species (2n = 6x = 60, 2C = 3,700 Mb) hybridized with different Spartina species introduced from the American coasts, which resulted in the formation of new invasive hybrids and allopolyploids. Thus, S. maritima raises evolutionary and ecological interests. However, genomic information is dramatically lacking in this genus. In an effort to develop genomic resources, we analysed 40,641 high-quality bacterial artificial chromosome-end sequences (BESs), representing 26.7 Mb of the S. maritima genome. BESs were searched for sequence homology against known databases. A fraction of 16.91 % of the BESs represents known repeats including a majority of long terminal repeat (LTR) retrotransposons (13.67 %). Non-LTR retrotransposons represent 0.75 %, DNA transposons 0.99 %, whereas small RNA, simple repeats and low-complexity sequences account for 1.38 % of the analysed BESs. In addition, 4,285 simple sequence repeats were detected. Using the coding sequence database of Sorghum bicolor, 6,809 BESs found homology accounting for 17.1 % of all BESs. Comparative genomics with related genera reveals that the microsynteny is better conserved with S. bicolor compared to other sequenced Poaceae, where 37.6 % of the paired matching BESs are correctly orientated on the chromosomes. We did not observe large macrosyntenic rearrangements using the mapping strategy employed. However, some regions appeared to have experienced rearrangements when comparing Spartina to Sorghum and to Oryza. This work represents the first overview of S. maritima genome regarding the respective coding and repetitive components. The syntenic relationships with other grass genomes examined here help clarifying evolution in Poaceae, S. maritima being a part of the poorly-known Chloridoideae sub-family.
Link
A high density physical map of chromosome 1BL supports evolutionary studies, map-based cloning and sequencing in wheat.
Genome Biol. 2013 Jun 25;14(6):R64. [Epub ahead of print]
Voir plus
Authors
Philippe R, Paux E, Bertin I, Sourdille P, Choulet F, Laugier C, Simkova H, Safar J, Bellec A, Vautrin S, Frenkel Z, Cattonaro F, Magni F, Scalabrin S, Martis MM, Mayer KF, Korol A, Berges H, Dolezel J, Feuillet C.
Genome Biol. 2013 Jun 25;14(6):R64. [Epub ahead of print]
Abstract
BACKGROUND:
As for other major crops, achieving a complete wheat genome sequence is essential for the application of genomics to breeding new and improved varieties. To overcome the complexities of the large, highly repetitive and hexaploid wheat genome, the International Wheat Genome Sequencing Consortium established a chromosome-based strategy that was validated by the construction of the physical map of chromosome 3B. Here, we present improved strategies for the construction of highly integrated and ordered wheat physical maps, using chromosome 1BL as a template, and illustrate their potential for evolutionary studies and map-based cloning.
RESULTS:
Using a combination of novel high throughput marker assays and an assembly program, we developed a high quality physical map representing 93% of wheat chromosome 1BL, anchored and ordered with 5,489 markers including 1,161 genes. Analysis of the gene space organization and evolution revealed that gene distribution and conservation along the chromosome results from the superimposition of the ancestral grass and recent wheat evolutionary patterns leading to a peak of synteny in the central part of the chromosome arm and an increased density of non collinear genes towards the telomere. With a density of about 11 markers per Mb, the 1BL physical map provides 916 markers, including 193 genes, for fine mapping the 40 QTLs mapped on this chromosome.
CONCLUSIONS:
Here, we demonstrate that high marker density physical maps can be developed in complex genomes such as wheat to accelerate map-based cloning, gain new insights into genome evolution, and provide a foundation for reference sequencing.:
MtQRRS1, an R-locus required for Medicago truncatula quantitative resistance to Ralstonia solanacearum.
New Phytol. 2013 May 2. doi: 10.1111/nph.12299.
Voir plus
Authors
Ben C, Debellé F, Berges H, Bellec A, Jardinaud MF, Anson P, Huguet T, Gentzbittel L, Vailleau F.
New Phytol. 2013 May 2. doi: 10.1111/nph.12299.
Abstract
Ralstonia solanacearum is a major soilborne pathogen that attacks > 200 plant species, including major crops. To characterize MtQRRS1, a major quantitative trait locus (QTL) for resistance towards this bacterium in the model legume Medicago truncatula, genetic and functional approaches were combined. QTL analyses together with disease scoring of heterogeneous inbred families were used to define the locus. The candidate region was studied by physical mapping using a bacterial artificial chromosome (BAC) library of the resistant line, and sequencing. In planta bacterial growth measurements, grafting experiments and gene expression analysis were performed to investigate the mechanisms by which this locus confers resistance to R. solanacearum. The MtQRRS1 locus was localized to the same position in two recombinant inbred line populations and was narrowed down to a 64 kb region. Comparison of parental line sequences revealed 15 candidate genes with sequence polymorphisms, but no evidence of differential gene expression upon infection. A role for the hypocotyl in resistance establishment was shown. These data indicate that the quantitative resistance to bacterial wilt conferred by MtQRRS1, which contains a cluster of seven R genes, is shared by different accessions and may act through intralocus interactions to promote resistance.
Physical Mapping Integrated with Syntenic Analysis to Characterize the Gene Space of the Long Arm of Wheat Chromosome 1A.
PLoS One. 2013 Apr 16;8(4):e59542. doi: 10.1371/journal.pone.0059542.
Voir plus
Authors
Lucas SJ, Akpınar BA, Kantar M, Weinstein Z, Aydınoğlu F, Safář J, Simková H, Frenkel Z, Korol A, Magni F, Cattonaro F, Vautrin S, Bellec A, Bergès H, Doležel J, Budak H.
PLoS One. 2013 Apr 16;8(4):e59542. doi: 10.1371/journal.pone.0059542.
Abstract
BACKGROUND:
Bread wheat (Triticum aestivum L.) is one of the most important crops worldwide and its production faces pressing challenges, the solution of which demands genome information. However, the large, highly repetitive hexaploid wheat genome has been considered intractable to standard sequencing approaches. Therefore the International Wheat Genome Sequencing Consortium (IWGSC) proposes to map and sequence the genome on a chromosome-by-chromosome basis.
METHODOLOGY PRINCIPAL FINDINGS:
We have constructed a physical map of the long arm of bread wheat chromosome 1A using chromosome-specific BAC libraries by High Information Content Fingerprinting (HICF). Two alternative methods (FPC and LTC) were used to assemble the fingerprints into a high-resolution physical map of the chromosome arm. A total of 365 molecular markers were added to the map, in addition to 1122 putative unique transcripts that were identified by microarray hybridization. The final map consists of 1180 FPC-based or 583 LTC-based contigs.
CONCLUSIONS SIGNIFICANCE:
The physical map presented here marks an important step forward in mapping of hexaploid bread wheat. The map is orders of magnitude more detailed than previously available maps of this chromosome, and the assignment of over a thousand putative expressed gene sequences to specific map locations will greatly assist future functional studies. This map will be an essential tool for future sequencing of and positional cloning within chromosome 1A.
Contrasted Patterns of Molecular Evolution in Dominant and Recessive Self-Incompatibility Haplotypes in Arabidopsis.
PLoS Genet.2012 Mar;8(3):e1002495.
PMID: 22457631
Voir plus
Authors
Goubet PM, Bergès H, Bellec A, Prat E, Helmstetter N, Mangenot S, Gallina S, Holl AC, Fobis-Loisy I, Vekemans X, Castric V.
PLoS Genet. 2012 Mar;8(3):e1002495.
PMID: 22457631
Abstract
Self-incompatibility has been considered by geneticists a model system for reproductive biology and balancing selection, but our understanding of the genetic basis and evolution of this molecular lock-and-key system has remained limited by the extreme level of sequence divergence among haplotypes, resulting in a lack of appropriate genomic sequences. In this study, we report and analyze the full sequence of eleven distinct haplotypes of the self-incompatibility locus (S-locus) in two closely related Arabidopsis species, obtained from individual BAC libraries. We use this extensive dataset to highlight sharply contrasted patterns of molecular evolution of each of the two genes controlling self-incompatibility themselves, as well as of the genomic region surrounding them. We find strong collinearity of the flanking regions among haplotypes on each side of the S-locus together with high levels of sequence similarity. In contrast, the S-locus region itself shows spectacularly deep gene genealogies, high variability in size and gene organization, as well as complete absence of sequence similarity in intergenic sequences and striking accumulation of transposable elements. Of particular interest, we demonstrate that dominant and recessive S-haplotypes experience sharply contrasted patterns of molecular evolution. Indeed, dominant haplotypes exhibit larger size and a much higher density of transposable elements, being matched only by that in the centromere. Overall, these properties highlight that the S-locus presents many striking similarities with other regions involved in the determination of mating-types, such as sex chromosomes in animals or in plants, or the mating-type locus in fungi and green algae.
The Medicago genome provides insight into the evolution of rhizobial symbioses.
Nature. 2011 Nov 16.
Voir plus
Authors
Young ND, Debellé F, Oldroyd GE, Geurts R, Cannon SB, Udvardi MK, Benedito VA, Mayer KF, Gouzy J, Schoof H, Van de Peer Y, Proost S, Cook DR, Meyers BC, Spannagl M, Cheung F, De Mita S, Krishnakumar V, Gundlach H, Zhou S, Mudge J, Bharti AK, Murray JD, Naoumkina MA, Rosen B, Silverstein KA, Tang H, Rombauts S, Zhao PX, Zhou P, Barbe V, Bardou P, Bechner M, Bellec A, Berger A, Bergès H, Bidwell S, Bisseling T, Choisne N, Couloux A, Denny R, Deshpande S, Dai X, Doyle JJ, Dudez AM, Farmer AD, Fouteau S, Franken C, Gibelin C, Gish J, Goldstein S, González AJ, Green PJ, Hallab A, Hartog M, Hua A, Humphray SJ, Jeong DH, Jing Y, Jöcker A, Kenton SM, Kim DJ, Klee K, Lai H, Lang C, Lin S, Macmil SL, Magdelenat G, Matthews L, McCorrison J, Monaghan EL, Mun JH, Najar FZ, Nicholson C, Noirot C, O'Bleness M, Paule CR, Poulain J, Prion F, Qin B, Qu C, Retzel EF, Riddle C, Sallet E, Samain S, Samson N, Sanders I, Saurat O, Scarpelli C, Schiex T, Segurens B, Severin AJ, Sherrier DJ, Shi R, Sims S, Singer SR, Sinharoy S, Sterck L, Viollet A, Wang BB, Wang K, Wang M, Wang X, Warfsmann J, Weissenbach J, White DD, White JD, Wiley GB, Wincker P, Xing Y, Yang L, Yao Z, Ying F, Zhai J, Zhou L, Zuber A, Dénarié J, Dixon RA, May GD, Schwartz DC, Rogers J, Quétier F, Town CD, Roe BA.
Nature. 2011 Nov 16.
Abstract
Legumes (Fabaceae or Leguminosae) are unique among cultivated plants for their ability to carry out endosymbiotic nitrogen fixation with rhizobial bacteria, a process that takes place in a specialized structure known as the nodule. Legumes belong to one of the two main groups of eurosids, the Fabidae, which includes most species capable of endosymbiotic nitrogen fixation. Legumes comprise several evolutionary lineages derived from a common ancestor 60 million years ago (Myr ago). Papilionoids are the largest clade, dating nearly to the origin of legumes and containing most cultivated species. Medicago truncatula is a long-established model for the study of legume biology. Here we describe the draft sequence of the M. truncatula euchromatin based on a recently completed BAC assembly supplemented with Illumina shotgun sequence, together capturing ∼94% of all M. truncatula genes. A whole-genome duplication (WGD) approximately 58 Myr ago had a major role in shaping the M. truncatula genome and thereby contributed to the evolution of endosymbiotic nitrogen fixation. Subsequent to the WGD, the M. truncatula genome experienced higher levels of rearrangement than two other sequenced legumes, Glycine max and Lotus japonicus. M. truncatula is a close relative of alfalfa (Medicago sativa), a widely cultivated crop with limited genomics tools and complex autotetraploid genetics. As such, the M. truncatula genome sequence provides significant opportunities to expand alfalfa's genomic toolbox.
A 3000-loci transcription map of chromosome 3B unravels the structural and functional features of gene islands in hexaploid wheat.
Plant Physiol. 2011 Oct 27.
Voir plus
Authors
Rustenholz C, Choulet F, Laugier C, Safár J, Simková H, Dolezel J, Magni F, Scalabrin S, Cattonaro F, Vautrin S, Bellec A, Bergès H, Feuillet C, Paux E.
Plant Physiol. 2011 Oct 27.
Abstract
To improve our understanding of the organization and regulation of the wheat (Triticum aestivum L.) gene space, we established the first transcription map of a wheat chromosome (3B) by hybridizing a newly developed wheat expression microarray with BAC pools from a new version of the 3B physical map as well as with cDNA probes derived from 15 RNA samples. Mapping data for almost 3000 genes showed that the gene space spans the whole chromosome 3B with a twofold increase of gene density towards the telomeres due to an increase in the number of genes in islands. Comparative analyses with rice and Brachypodium revealed that these gene islands are composed mainly of genes likely originating from interchromosomal gene duplications. Gene ontology and expression profile analyses for the 3000 genes located along the chromosome revealed that the gene islands are enriched significantly in genes sharing the same function or expression profile, thereby suggesting that genes in islands acquired shared regulation during evolution. Only a small fraction of these clusters of cofunctional and coexpressed genes was conserved with rice and Brachypodium indicating a recent origin. Finally, genes with the same expression profiles in remote islands (coregulation islands) were identified suggesting long-distance regulation of gene expression along the chromosomes in wheat.
Functional features of a single chromosome arm in wheat (1AL) determined from its structure.
Funct Integr Genomics. 2011 Sep 3.
Voir plus
Authors :
Lucas SJ, Simková H, Safár J, Jurman I, Cattonaro F, Vautrin S, Bellec A, Berges H, Doležel J, Budak H.
Funct Integr Genomics. 2011 Sep 3.
Abstract :
Bread wheat (Triticum aestivum L.) is one of the most important crops globally and a high priority for genetic improvement, but its large and complex genome has been seen as intractable to whole genome sequencing. Isolation of individual wheat chromosome arms has facilitated large-scale sequence analyses. However, so far there is no such survey of sequences from the A genome of wheat. Greater understanding of an A chromosome could facilitate wheat improvement and future sequencing of the entire genome. We have constructed BAC library from the long arm of T. aestivum chromosome 1A (1AL) and obtained BAC end sequences from 7,470 clones encompassing the arm. We obtained 13,445 (89.99%) useful sequences with a cumulative length of
7.57 Mb, representing 1.43% of 1AL and about 0.14% of the entire A genome. The GC content of the sequences was 44.7%, and 90% of the chromosome was estimated to comprise repeat sequences, while just over 1% encoded expressed genes. From the sequence data, we identified a large number of sites suitable for development of molecular markers
(362 SSR and 6,948 ISBP) which will have utility for mapping this chromosome and for marker assisted breeding. From 44 putative ISBP markers tested 23 (52.3%) were found to be useful. The BAC end sequence data also enabled the identification of genes and syntenic blocks specific to chromosome 1AL, suggesting regions of particular functional interest and targets for future research.
Construction and characterization of two BAC libraries representing a deep-coverage of the genome of chicory (Cichorium intybus L., Asteraceae).
BMC Res Notes. 2010 Aug 11;3:225.
Voir plus
Abstract
BACKGROUND: The Asteraceae represents an important plant family with respect to the numbers of species present in the wild and used by man. Nonetheless, genomic resources for Asteraceae species are relatively underdeveloped, hampering within species genetic studies as well as comparative genomics studies at the family level. So far, six BAC libraries have been described for the main crops of the family, i.e. lettuce and sunflower. Here we present the characterization of BAC libraries of chicory (Cichorium intybus L.) constructed from two genotypes differing in traits related to sexual and vegetative reproduction. Resolving the molecular mechanisms underlying traits controlling the reproductive system of chicory is a key determinant for hybrid development, and more generally will provide new insights into these traits, which are poorly investigated so far at the molecular level in Asteraceae.
FINDINGS: Two bacterial artificial chromosome (BAC) libraries, CinS2S2 and CinS1S4, were constructed from HindIII-digested high molecular weight DNA of the contrasting genotypes C15 and C30.01, respectively. C15 was hermaphrodite, non-embryogenic, and S2S2 for the S-locus implicated in self-incompatibility, whereas C30.01 was male sterile, embryogenic, and S1S4. The CinS2S2 and CinS1S4 libraries contain 89,088 and 81,408 clones. Mean insert sizes of the CinS2S2 and CinS1S4 clones are 90 and 120 kb, respectively, and provide together a coverage of 12.3 haploid genome equivalents. Contamination with mitochondrial and chloroplast DNA sequences was evaluated with four mitochondrial and four chloroplast specific probes, and was estimated to be 0.024% and 1.00% for the CinS2S2 library, and 0.028% and 2.35% for the CinS1S4 library. Using two single copy genes putatively implicated in somatic embryogenesis, screening of both libraries resulted in detection of 12 and 13 positive clones for each gene, in accordance with expected numbers.
CONCLUSIONS: This indicated that both BAC libraries are valuable tools for molecular studies in chicory, one goal being the positional cloning of the S-locus in this Asteraceae species.
Authors
Gonthier L, Bellec A, Blassiau C, Prat E, Helmstetter N, Rambaud C, Huss B, Hendriks T, Bergès H, Quillet MC.
BMC Res Notes. 2010 Aug 11;3:225.