

- Project coordinator : William Marande
- Project Partners
- Funding: FRAIB
- CNRGV's responsible : Isabelle Dufau
- CNRGV implactions: HWM-DNA extraction, Optical Map
- Vincent Castric
- Isabelle Loisy - Thierry Gaude
- Jacinthe Azevedo - Thierry Lagrange
- Establish a high quality reference genome sequence for Quercus robur (740Mb/C), anchored to an ultra high density SNP-based linkage map on which a series of trait-QTLs have been localized. This resource will provide genetic links between traits and sequence variations at a high resolution,
- Identify genes involved in adaptation through the analysis of adaptive divergence (incipient ecological speciation) at the transcriptome and genome-wide levels by respectively i) identifying differentially expressed genes between co-occuring species (sessile and pedunculate oaks) adapted to different edapho-climatic environments, and ii) analyzing the size, distribution and nature of genomic islands of divergence maintained in the face of gene flow of the same sympatric species.
- Obtain a reference genome for a single Pedunculate oak genotype
- Assign genome scaffolds on the genome using a highly saturated SNP-based genetic linkage map
- Annotate structurally and functionally the genome
- Integrate the whole genome with automatic prediction into a Genome Browser
- Study the micro-synteny and colinearity between oak and chestnut, as well as with other eudicots, for which a genome sequence is available
- Reveal genetic variations that are responsible for important adaptive traits in oaks by resequencing the genome of several individuals of two sister species.
- https://w3.pierroton.inra.fr/QuercusPortal/index.php?p=OAK_GENOME_SEQUENCING
- http://www.oakgenome.fr/
- Oak genome reveals facets of long lifespan.
- Decoding the oak genome: public release of sequence data, assembly, annotation and publication strategies.
- Analysis of BAC end sequences in oak, a keystone forest tree species, providing insight into the composition of its genome.
- William Marande
- William Marande
- Construction and screening of BAC libraries
- BAC clone characterization
-
Dr. Eric Jenczewski
Institut Jean-Pierre Bourgin
Centre INRA Versailles-Grignon
RD10
78026 Versailles
Email : eric.jenczewski@versailles.inra.fr -
INRA Site de Crouël
UMR1095 GDEC Génétique Diversité et Ecophysiologie des Céréales
234 avenue du Brézet
63100 CLERMONT-FERRAND
Scientist in charge of the project : Pierre Sourdille
Email : pierre.sourdille@clermont.inra.fr
- William Marande

gEXTRACT
The perennial legume Mimosa pudica fits in that category as it is a recalcitrant plant producing high amounts of secondary metabolites and polysaccharides...
Voir plus
Since the Next Generation Sequencing revolution in 2005, whole genomes sequencing has become a routine activity for all types of species and for multiple research.
The main limitation to take full advantage of long read sequencing and scaffolding technologies is the need for High Molecular Weight (HMW) DNA as starting material.
While many protocols to obtain HMW DNA have been developed for mammals only few methods dedicated to plants exist and the quality of DNA obtained rarely meets the quality required for successful sequencing.
The perennial legume Mimosa pudica fits in that category as it is a recalcitrant plant producing high amounts of secondary metabolites and polysaccharides.
M. pudica is however of particular interest for several reasons : model for the study of mechanosensing systems in plants, formation of the nitrogen-fixing root nodule symbiosis (RNS), representative model of the legume subfamily Mimosoideae.
A gold-standard M. pudica genome assembly is required, but multiple protocols tested for DNA extraction finally resulted in short fragment only suitable for Illumina-based sequencing.
Failing to extract HMW gDNA for long-read sequencing is not specific to M. pudica, so the main goal of the gEXTRACT project will be to leverage the expertise of our three labs (CNRGV, LIPM, LRSV) to develop a universal protocol for plant HMW gDNA extraction.
This protocol will be an essential resource for any laboratories interested in plant genome sequencing.
The project is divided in three objectives:
1) Optimisation of a HMW gDNA extraction protocol suitable for long-read sequencing on M. pudica samples
2) Generation of a gold-standard genomic assembly for Mimosa pudica using PacBio (long-read sequencing) and Bionano (optical mapping).
3) Extension of the HMW gDNA extraction protocol to a diversity of plant species (ex: the liverworts Marchantia paleacea, the fern Pteris vittata, the non-RNS forming legume Chaetocalyx brasilense, the Zygnematophyceae Spirogloea muscicola) to demonstrate the universal efficiency of the gEXTRACT protocol.
24 Chemin de Borde Rouge
31320 Castanet tolosan
24 Chemin de Borde Rouge
31320 Castanet tolosan

Genomic analyses reveal of self-incompatibility in Capsella
This project aims at characterizing the genetic basis and timing of loss of SI in the self-fertilizing crucifer species Capsella orientalis using complementary approaches (long read sequencing of multiple full-length S-haplotypes, genetic mapping, population genomics and expression analyses).
Voir plus
This project aims at characterizing the genetic basis and timing of loss of SI in the self-fertilizing crucifer species Capsella orientalis using complementary approaches (long read sequencing of multiple full-length S-haplotypes, genetic mapping, population genomics and expression analyses).
Plants have many intricate mechanisms to promote outcrossing. A common mechanism is self-incompatibility (SI), which allows recognition and rejection of self pollen through male and female specificity components, encoded at the S-locus. Despite the benefits of outcrossing, SI has repeatedly been lost. Theory predicts that mutations disrupting SI spread at different paces depending on whether they affect male or female functions, involve unlinked modifiers and whether they are dominant or recessive, but the importance of parallel changes for loss of SI remains unclear. The crucifer genus Capsella offers an excellent opportunity to study multiple transitions from outcrossing to self-fertilization, but so far, little is known about the genetic basis and timing of loss of SI in the self-fertilizing diploid Capsella orientalis.
Collaborators
Tanja Slotte: Department of Ecology, Environment and Plant Sciences, Science for Life Laboratory, Stockholm University, SE-106 91 Stockholm, Sweden,
ClaudiaKöhler : Department of Plant Biology, Swedish University of Agricultural Sciences & Linnean Center for Plant Biology, SE-75 007 Uppsala, Sweden,
Vincent Castric Unité Evo-Eco-Paléo (EEP) - UMR 8198, CNRS/Université de Lille - Sciences et Technologies, Villeneuve d'Ascq Cedex, F-59655, France, 5Equal contributions
Responsible
William Marande, Caroline Callot

CONSORTIUM for the sequencing of Vanilla planifolia - « VANISEQ » project
This consortium aims at providing genomic resources and data in order to better understand the genetic of Vanilla planifolia.
Vanilla is a very popular flavoring material. Of the approximately 110 Vanilla species (Orchidaceae, monocot) that have been identified, only two are important in terms of commerce and cultivation; V. planifolia Andrews and V. tahitensis JW Moore. Among these, V. planifolia is the most valued for its flavor qualities and is therefore widely cultivated and used for the production of food additives.
Voir plus
This consortium aims at providing genomic resources and data in order to better understand the genetic of Vanilla planifolia.
Vanilla is a very popular flavoring material. Of the approximately 110 Vanilla species (Orchidaceae, monocot) that have been identified, only two are important in terms of commerce and cultivation; V. planifolia Andrews and V. tahitensis JW Moore. Among these, V. planifolia is the most valued for its flavor qualities and is therefore widely cultivated and used for the production of food additives. The fully-grown mature fruits of vanilla, also called beans or pods, develop characteristic aromatic properties by the process of curing. The cured beans are referred to as vanilla, and the major flavor compound is vanillin (3-methoxy, 4-hydroxy-benzaldehyde). In spite of its economic importance, there is little genetic diversity and few genetic or genomic resources in V. planifolia (Rao et al., 2014). The chromosome number for V. planifolia has been reported as 2n = 32. Most V. planifolia accessions are considered to be diploid with a 2C-value of 5.03 pg, but due to the large size and complexity of the V. planifolia genome, limited sequence resources are currently available.
Rao X, Krom N, Tang Y, Widiez T, Havkin-Frenkel D, Belanger FC, Dixon RA, Chen F. (2014). A deep transcriptomic analysis of pod development in the vanilla orchid (Vanilla planifolia). BMC Genomics. 15:964. doi: 10.1186/1471-2164-15-964.
Collaborators
CIRAD and Université de la Réunion
Michel GRISONI
Pôle de protection des plantes, 7 chemin de l’IRAT, 97410 Saint-Pierre, Réunion
+262 2 62 49 92 28
michel.grisoni@cirad.fr
MANE
Joseph ZUCCA
V MANE FILS Département Biotechnologies 620 route de Grasse 06620 Le Bar Sur Loup
04 96 09 75 91
joseph.zucca@mane.com
EUROVANILLE
Antoine DU GRANRUT
Route de Maresquel 62879 Gouy-Saint-André
+33 (0)6 35 21 39 50
adugranrut@eurovanille.com
EVT
Sandra LEPERS
BP 912 98735 Uturoa Polynésie française
+689 40 66 46 59
sandra.lepers@vanilledetahiti.fr
INRA
Dr Helene BERGES
Director of the French Plant Genomic Resource Center
Chemin de Borde Rouge CS 52627
31326 Castanet Tolosan cedex
Tél. : 33(0) 561 28 55 65
Fax : 33(0) 561 28 55 64
Mobile : +33 (0)6 07 95 89
Helene.Berges@inra.fr
Responsible
William MARANDE

Examining the history and impact of gene content variation in sunflower
The proposed research will contribute to new resources for the sunflower genomics community including new draft genome assemblies and annotations for wild Helianthus annuus and an early domesticate. We will also test our hypothesis that copy number variation has been an important contributor to sunflower domestication, and by combining this knowledge with our gene expression datasets and published trait mapping datasets, we may highlight variants useful for further improvement of agronomic traits like yield, oil content, and stress resistance. Finally, this project will test whether the applications of optical mapping can be extended to identifying large chromosomal variants that distinguish pools of DNAs.
Voir plus
The proposed research will contribute to new resources for the sunflower genomics community including new draft genome assemblies and annotations for wild Helianthus annuus and an early domesticate. We will also test our hypothesis that copy number variation has been an important contributor to sunflower domestication, and by combining this knowledge with our gene expression datasets and published trait mapping datasets, we may highlight variants useful for further improvement of agronomic traits like yield, oil content, and stress resistance. Finally, this project will test whether the applications of optical mapping can be extended to identifying large chromosomal variants that distinguish pools of DNAs. In addition to the use of this approach to examine the historical action of divergent selection on these variants as proposed here, this application could also be applied for bulk segregant mapping of trait variation in crosses developed from contemporary germplasm. Finally, the collaboration will train junior scientists, enrich ongoing and future research projects by the collaborating labs, and expand the range and quality of services of the primary French facility for agricultural genomics.
One of the most impactful insights gained from the onslaught of sequence data being generated for plant genomes using high-throughput genomic sequencing technologies is a single individual’s genome may contain just half of the total gene set present across all individuals of the species as a whole. In other words, a substantial portion of the genes present in one individual’s genome may be absent from the genome of another individual or vice versa. Consequently, the pan-genome, or the total functional gene sequence present among all individuals of a species, is far larger than the content annotated in a single reference genome. These gene presence/absence polymorphisms segregating among individuals within species arise through large duplication, insertion, or deletion events and can be important sources of variation that have contributed in the past to crop domestication or adaptation of wild populations to local habitats. Moreover, this form of variation is also a critical source of genetic diversity to draw upon in the future to improve crop yields in the face of changing climates, especially as losses of diversity during the history of cultivation may have disproportionately removed variants favorable under stressful environments from the gene pool. Here we propose a collaboration to develop advanced genomic resources for wild and domesticated accessions of the common sunflower, Helianthus annuus, to facilitate greater understanding of the role of gene content variation in its history and breeding potential.
Collaborators
Dr. Benjamin Blackman - University of California, Berkeley (UC Berkeley) - 111 Koshland Hall #3102 - Berkeley, CA 94720 - United States
http://plantandmicrobiology.berkeley.edu/profile/bblackman
https://nature.berkeley.edu/blackmanlab/Blackman_Lab/Welcome.html
Responsible
William Marande

Analysis of WRKY and ERF genes potentially Involved in salt stress responses in Triticum turgidum L. ssp. durum.
WRKY transcription factors are involved in multiple aspects of plant growth, development and responses to biotic stresses. Although they have been found to play roles in regulating plant responses to environmental stresses, these roles still need to be explored, especially those pertaining to crops. Durum wheat is the second most widely produced cereal in the world. Complex, large and unsequenced genomes, in addition to a lack of genomic resources, hinder the molecular characterization of tolerance mechanisms. This project aims at characterizing TdWRKY genes from durum wheat (Triticum turgidum L. ssp. durum).
Voir plus
WRKY transcription factors are involved in multiple aspects of plant growth, development and responses to biotic stresses. Although they have been found to play roles in regulating plant responses to environmental stresses, these roles still need to be explored, especially those pertaining to crops. Durum wheat is the second most widely produced cereal in the world. Complex, large and unsequenced genomes, in addition to a lack of genomic resources, hinder the molecular characterization of tolerance mechanisms. This project aims at characterizing TdWRKY genes from durum wheat (Triticum turgidum L. ssp. durum). Enrichment of cis-regulatory elements related to stress responses in the promoters of some TdWRKY genes indicated their potential roles in mediating plant responses to a wide variety of environmental stresses. TdWRKY genes displayed different expression patterns in response to salt stress that distinguishes two durum wheat genotypes with contrasting salt stress tolerance phenotypes. The work done in collaboration indicates that these genes might play some functional role in the salt tolerance in durum wheat.
Collaborators
Laboratory of Plant Molecular Physiology, Center of Biotechnology of Borj Cedria, Borj Cedria Science and Technology ParkHammam-lif, Tunisia - Yousfi Fatma, Makhloufi Emna
CNRGV's responsible
William Marande
Publications related to the project

NOVEL: Emergence of novel phenotypes in co-evolving biological systems: allelic diversification and dominance at the Self-incompatibility locus in Arabidopsis.
ERC Consolidator Grant (CoG), LS8, ERC-2014-CoG
Voir plus
Project coordinator:
Unité Evo-Eco-Paléo (EEP) - UMR 8198
CNRS / Université de Lille - Sciences et Technologies
Batiment SN2, bureau 207
59655 Villeneuve d'Ascq - FRANCE
Project partners:
Laboratory « Reproduction et Developpement des Plantes »
ENS Lyon CNRS - France
Laboratory « Genome et Developpement des Plantes »
UMR 5096 (UPVD/CNRS) - Perpignan - France
Website:
http://eep.univ-lille.fr/fr/perso-vincent-castric
Abstract:
The emerging field of systems biology is revealing the intricate nature of biological organisms, whereby a large fraction of their individual components (genes, proteins, regulatory elements) interact with several others. The co-evolutionary processes that this entails raises the question of how phenotypic novelty may arise in the course of evolution, since all parts of the system have to evolve in a coordinated manner if the phenotype is to remain functional. For most biological systems, however, we are lacking even basic insight into the fine-scale mechanistic constraints and the underlying ecological context. In this project, we will focus on the sporophytic self-incompatibility system in outcrossing Arabidopsis species, a model biological system in which two distinct co-evolutionary processes are becoming well-understood: 1) between the male and female reproductive proteins allowing self-pollen recognition and rejection and 2) between small non-coding RNAs and their target sites that jointly control the dominance/recessivity interactions between self-incompatibility alleles. By studying these two model systems, we will aim to catch the emergence of functional and regulatory novelty in flagrante delicto. We will take a multidisciplinary approach combining theoretical and empirical population genetics, evolutionary genomics and ancestral protein resurrection using transgenic plants. Our goal is threefold: 1) decrypt the molecular alphabet of the interaction between co-evolving nucleotide sequences, 2) predict and evaluate the fitness landscapes upon which the two co-evolutionary processes are taking place and 3) exploit natural variation in closely related species to unveil the kind of co-evolutionary process in natural populations. Our combination of various powerful approaches in a tractable model system should provide insight on diversification, a poorly understood but fundamental evolutionary process that is taking place at all levels of organization.
This project is an extension of the project BRASSIDOM
| CNRGV's involvement: BAC library construction and screening
CNRGV's responsible: William Marande |
|
Publications related to the project:
The unusual S locus of Leavenworthia is composed of two sets of paralogous loci

ERC LUPIN ROOTS : Unravelling cluster root development in white lupin
Plants exhibit a high level of developmental plasticity that is controlled by a complex combination of perception, integration and response. In opposition to animals, where developmental patterns are highly conserved, plant organs are produced as a response to environmental stimuli. Understanding the molecular mechanisms involved in how plants perceive and respond to these stimuli is of key importance and may lead to future application in the agronomic field. One of the most striking developmental adaptations are cluster roots from White Lupin (Lupinus albus)...
Voir plus
Plants exhibit a high level of developmental plasticity that is controlled by a complex combination of perception, integration and response. In opposition to animals, where developmental patterns are highly conserved, plant organs are produced as a response to environmental stimuli. Understanding the molecular mechanisms involved in how plants perceive and respond to these stimuli is of key importance and may lead to future application in the agronomic field. One of the most striking developmental adaptations are cluster roots from White Lupin (Lupinus albus). Cluster roots are produced in response to low phosphate and are made of hundreds of closely spaced lateral roots that harbor a highly specialized development, growth, differentiation and physiology. They represent a fantastic tool to study root plant developmental adaptation.
Led by Benjamin Péret, CNRS researcher and team leader of Plasticity Team at BPMP, the "LUPIN ROOTS" ERC Starting grant project will develop tools to study cluster roots and make white lupin a good model. We will sequence the genome, develop genetic transformation, produce a transcriptomic database of the cluster roots and a mutagenised population to retrieve mutants affected in cluster root development. This project will lead to a better understanding of how plants are able to perceive their environment and modify their development accordingly. In the future, this will help to diminish the amount of phosphate fertilizer used to grow crops.
|
| This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement No 637420). |
Project coordinator
Benjamin PERET
CNRS
UMR BPMP
2 Place Pierre Viala
34060 MONTPELLIER
Email: benjamin.peret@cnrs.fr
Website: https://www1.montpellier.inra.fr/wp-inra/bpmp/en/research/the-teams/development-and-plasticity-of-the-root-system/
CNRGV involvement:
The CNRGV will coordinate the genome sequencing project and will produce an optical map using the Irys system from Bionano genomic to improve the genome assembly. The CNRGV will produce the high molecular weight DNA for the PacBio and Oxford Nanopore sequencing.
Responsible: William Marande
Collaborators
Jérôme Gouzy (Bio-informatic platform, INRA - LIPM)

Identifying sequences associated with virus tolerance in cassava
Geminiviruses represent an important threat to crop production and food security as they are the causal agents of viral diseases in tropical and temperate staple crops. Understanding natural host resistance opens new perspectives to reduce the negative impact of viral diseases on crop production...
Voir plus
Geminiviruses represent an important threat to crop production and food security as they are the causal agents of viral diseases in tropical and temperate staple crops. Understanding natural host resistance opens new perspectives to reduce the negative impact of viral diseases on crop production.
Cassava geminiviruses (CGMs) are the causal agents of cassava mosaic disease (CMD), the most widespread and devastating disease of cassava. Natural host tolerance against CGMs has been identified in the cassava germplasm and extensively used in order to breed CMD tolerant varieties. Despite the important use of the so-called CMD2-based dominant monogenic resistance, tolerance mechanisms have not yet been characterized in cassava.
A whole genome and targeted genome sequencing approach have been used to determine the genomic structure of the CMD2 locus. Despite a relatively good resolution in the CMD2 locus, the project will further increase the resolution by BAC sequencing approach. BAC sequencing approach will contribute to the precise characterization of the polymorphism in the CMD2 region of cassava accessions contrasting for geminivirus tolerance.
Project coordinator:
Plant Genetics Lab, AgroBioChem Department, Gembloux Agro Bio-Tech,
University of Liège, Belgium
Bâtiment 48 N-1 // Room 48-01-40
Avenue Maréchal Juin
5030 Gembloux
Belgium
Represented by Pr Herve Vanderschuren
Email: herve.vanderschuren@ulg.ac.be
Tel: +32 81 62 25 71
Website: http://www.gembloux.ulg.ac.be/plant-genetics/
CNRGV involvement:
Responsible: Caroline Callot and William Marande
BAC libraires construction – screening – BAC clone sequencing

GENOAK : Sequencing of the oak genome and identification of genes that matter for forest tree adaptation
The objective of the GENOAK project are to establish a high quality reference genome sequence for Quercus robur, and to identify genes involved in adaptation through the analysis of adaptive divergence (incipient ecological speciation) at the transcriptome and genome-wide levels.
Voir plus
Trees are an important component of the global ecosystem. They dominate many regions of the world as natural, extensively or intensively managed plantations. The Fagaceae family comprises about 1,000 woody species distributed throughout the northern hemisphere. About half belong to the Quercus (oak) genus. These oaks have traditionally high cultural and societal values. As other forest tree species, they are also providing important environmental (carbon sequestration, water cycle, reservoir of biodiversity, soil protection...) and economic (carpentry, furniture, cabinet making, veneer, cask industry, fuelwood, hunting and fungus gathering) services.
Finally these long-lived organisms are considered as relevant models to study both short and long term adaptation mechanisms to abiotic and biotic constraints associated with global change, as they grow under a wide range of environments (soil and climate). Due to their biological dominance, their commercial and ecological importance worldwide, an international network was launched recently to develop a coordinated strategy to sequence several Fagaceae genomes. These genomic resources will allow to discover which genes are involved in the adaptation of these organisms to their environments and to study their adaptive potential under current and future climate changes.
In Europe, genomic resources have been developed for sessile and pedunculate oaks in the frame of the EVOLTREE network of excellence. In US, American and Chinese chestnuts (Castanea) were the main targets of a NSF funded project. Their genome is now being sequenced within the Forest Health Initiative . The goal is to provide a high-quality reference sequence for the Castanea mollissima (Chinese Chestnut) genome, with which to identify genes for resistance to Cryphonectria parasitica. In China, the efforts to sequence the Lithocarpus and Castanopsis genomes are also on the right track.
The present project aims at supporting the participation of French Institutions involved in this international effort. It is a collaboration between four INRA research units and the GENOSCOPE: the French National Sequencing Center.
The objective of the GENOAK project are two-fold:
Tasks :
Website :
Publications related to the project :
CNRGV's responsible
AMAIZING
AMAIZING aims at the development of innovative breakthrough in breeding methods and agricultural practices for the production of high yielding crop varieties with improved environmental values. It relies on a large partnership between the key players of maize economy in France and in particular a strong private partnership unique to date in the history of maize research in France.
Voir plus
Maize is currently the only major hybrid cereal crop in Europe that offers outstanding possibilities in terms of performance and environmental impact.French breeders are ranked second behind the USA in terms of maize seed production. Maize has demonstrated sustained gain in productivity in a global stagnation context in France.
In order to face the challenges of delivering safe and high-quality food and feed in a sustainable manner while maintaining yield and stability across different environments affected by climatic change, a paradigm shift is needed in maize breeding. Scientific breakthroughs are needed to take advantage of emerging technologies and better valorize the potential of this species.
AMAIZING aims at the development of innovative breakthrough in breeding methods and agricultural practices for the production of high yielding crop varieties with improved environmental values. It relies on a large partnership between the key players of maize economy in France and in particular a strong private partnership unique to date in the history of maize research in France.
CNRGV's responsible

Positional cloning of the SrWLR locus involved in the wheat stem rust resistance
The project is developed in collaboration with the laboratory of Dr Maricellis Acevedo (North Dakota State University) as part of Jason Zurn PhD thesis.
The project's objective is to characterize the SrWLR locus, responsible for stem rust resistance in wheat.
Voir plus
Project coordinator:
Dr Maricelis Acevedo and Jason Zurn (PhD student)
Department of Plant Pathology
North Dakota State University
Walster Hall, 306
Fargo, ND, USA 58102
Website:
http://www.ag.ndsu.edu/plantpath
Abstract:
The project is developed in collaboration with the laboratory of Dr Maricellis Acevedo (North Dakota State University) as part of Jason Zurn PhD thesis. The project's objective is to characterize the SrWLR locus, responsible for stem rust resistance in wheat.
In previous experiments the SrWLR locus has been positioned on chromosome 2BL and genetically mapped to a 0.38 cM region. In order to find candidates genes for this phenotype, we’ll establish a physical map of the region.
This will be done using the BAC library approach. We’ll create a BAC library from the resistant Iranian landrace PI 626573-2 and screen for markers located in the region targeted.
Publication:
High-density mapping of a resistance gene to Ug99 from the Iranian landrace PI 626573
JD Zurn, M Newcomb, MN Rouse, Y Jin, S Chao, J Sthapit, DR See, ...
Molecular Breeding 34 (2), 871-881
CNRGV involvement:
Responsible: William Marande
Construction and screening of a BAC library
Physical map of the genomic region of interest
Controling Recombination rate for pOlyploid Crop improvement (CROC)
Controling Recombination rate for pOlyploid Crop improvement (CROC)
Voir plus
Meiotic recombination is a fundamental process for all sexual eukaryotes; it is required to produce balanced gametes and therefore is essential to the fertility of species. Furthermore, meiotic recombination is also crucial for plant breeding because it allows, through the formation of crossovers (COs), to reshuffle genetic material between individuals and between species. Major international efforts have been made to identify the genes that are involved in meiotic recombination in plants, primarily using diploid Arabidopsis thaliana as model system. Therefore much of this work has disregarded the consequences of polyploidy, one of the key features of crop plant genomes, on meiotic recombination.Essential questions thus remain unsolved: How is meiotic recombination regulated in polyploid (crop) species? Why and how does polyploidy increase the rate of meiotic recombination? How can such improved knowledge on recombination be exploited for crop improvement? This project will address these questions specifically, using two complementary polyploid crop species: oilseed rape (Brassica napus; AACC; 2n=38) and bread wheat (Triticum aestivum; AABBDD; 2n=42).
We will set up a complete set of integrated analyses to explore many inter-related aspects of CO regulation in polyploid crops.
Task 1 aims at characterizing the molecular underpinnings of CO suppression between homeologous chromosomes in wheat and oilseed rape. We will proceed with positional cloning of the PrBn (in oilseed rape) and Ph2 (in wheat) loci. For this latter case, particular emphasis will be placed on evaluating TaMSH7, the most promising candidate for Ph2. CROC will thus advance understanding of the mechanisms that hamper the incorporation of beneficial traits from wild relatives into crop plants by promoting a diploid-like meiosis in allopolyploids; overcoming this specific stumbling block would openthe road to the creation of new crop varieties resistant to diseases and more efficient in nitrogen use (to name only these).
Task 2 will advance understanding on the cause of the striking CO rate increase we have discovered in Brassica
digenomic triploid AAC hybrid and its possible application to wheat. We will determine whether these extra COs i) arise from one or the other CO pathways and ii) can be combined with those resulting from the mutation of an antirecombination meiotic protein. We will unravel the individual and interaction effects of three C chromosomes on the rate and distribution of COs between homologues and test whether wheat pentaploid AABBD hybrids have the same boosting effect on CO frequencies as Brassica AAC triploid hybrids. The expected outcomes will pave the way to broaden the genetic variation that is available to plant breeders.
CROC is a timely project that is shaped to address fundamental questions with practical objectives; it is directly upstream of research on innovative plant breeding technologies contributing to the competitiveness of French/European Agriculture and thus completely relevant to this call. CROC combines a group of researchers with a comprehensive and complementary expertise and set of facilities. Its strong translational emphasis ensures that the results obtained will have general significance that extends beyond oilseed rape and bread wheat. Our work will thus shed new light on the pending cause of CO variation in polyploid plant species, a critical issue for genetics, evolution and plant breeding.
ANR Project
Project coordinator:
INRA IJPB
Eric Jenczewski
Route de Saint Cyr - RD10
78026 Versailles cedex
Project partner:
INRA GDEC
Pierre Sourdille
Chemin de Beaulieu
Domaine de Crouël
63039 Clermont-Ferrand
UMR1349 IGEPP
Anne Marie Chèvre
Domaine de la Motte
INRA BP35327
35653 Le Rheu
INRA EPGV
Marie-Christine Le Paslier
rue Gaston Crémieux
CEA-IG/CNG, Bat G2, CP5724
91057 Evry
INRA-CNRGV
Hélène Berges
Chemin de Borde Rouge
31326 Castanet Tolosan
CNRGV's responsible : William Marande
CNRGV involvement:
Funded by ANR (The French National Research Agency): ANR CROC
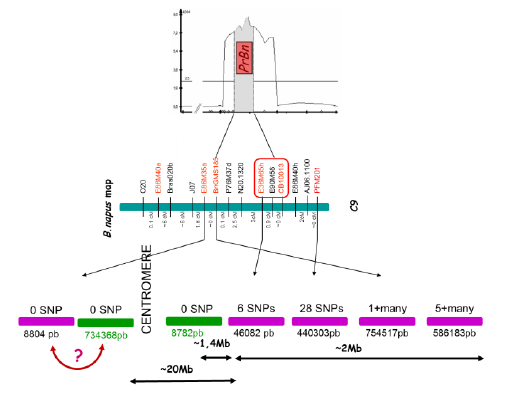
PrBn is the main determinant for CO formation between homeologous chromosomes in B. napus allohaploids; PrBn was first identified by conventional QTL analysis and mapped on chromosome C9, within a less than 10cM interval.
Using markers closely linked to PrBn as BLAST queries against the B. napus pseudomolecules, it was estimated that the entire region surrounding PrBn is ~20 Mb long and that it encompasses the centromere of C9. All but one of the markers most tightly linked to PrBn are located in an interval of ~2-3Mb on one side of the centromere that anchors 5-6 almost contiguous scaffolds. These markers are less than 2 Mb away from a meiotic recombination gene we mapped on C9.
Publication related to the project

Genetic Study of Barley Germplasm and Analysis of Segregating Generations of Cultivated and Wild Barley Crosses Under Drought Stress Conditions
Genetic Study of Barley Germplasm and Analysis of Segregating Generations of Cultivated and Wild Barley Crosses Under Drought Stress Conditions
Voir plus
Project coordinator:
Pr Mohammad Mahdi Majidi
Dr Aghafakhr Mirlohi
Mohammad Barati (PhD student)
Department of Agronomy and Plant Breeding
Collage of Agriculture
Isfahan University of Technology
Isfahan
Iran
Abstract:
Barley (Hordeum vulgare ssp. vulgare) is the 4th most important cereal. Wild barley (H. vulgare ssp. spontaneum), the progenitor of cultivated barley is originated from Fertile Crescent and is adapted to drought, the most important abiotic stress limiting crop production in the world and Iran. The project aimed at studying the genetic diversity of cultivated and wild barley under drought stress and transfer of favorite alleles from wild barley to cultivated varieties. The project consists of four studies. 1) Assessment of genetic variation for agro-morphological and root-related traits under drought stressed conditions. 2) Creation and evaluation of segregating populations. 3) Mapping chromosome regions controlling traits involved in drought tolerance. 4) Characterization of transcription factor families in cultivated and wild barley.
CNRGV involvement:
Responsible: William Marande
Identification and characterization of WRKY and DREB transcription factors in barley varieties using non-gridded BAC library approach

Ger4 locus characterization in 8 barley varieties and it relation with powdery mildew resistance
Ger4 locus characterization in 8 barley varieties and it relation with powdery mildew resistance
Voir plus
Project coordinator:
Patrick Schweizer, Dr.
Head of Genome Analysis program
Head of Pathogen-Stress Genomics lab
Leibniz-Institut für Pflanzengenetik und Kulturpflanzenforschung (IPK)
Genomzentrum
Corrensstrasse 3
D-06466 Gatersleben
Germany
Abstract:
Immunity of plants triggered by pathogen-associated molecular patterns (PAMPs) is based on the execution of an evolutionarily conserved defense response that includes the accumulation of pathogenesis-related (PR) proteins as well as multiple other defenses. The most abundant PR transcript of barley (Hordeum vulgare) leaf epidermis attacked by the powdery mildew fungus Blumeria graminis f. sp hordei encodes the germin-like protein GER4, which has superoxide dismutase activity and functions in PAMP-triggered immunity.
Patrick Schweizer ‘s team (IPK- Germany) showed that barley GER4 is encoded by a dense cluster of tandemly duplicated genes (GER4a-h) that underwent several cycles of duplication. The genomic organization of the GER4 locus also provides evidence for repeated gene birth and death cycles. They also identify 8 barley varieties that show different GER4 expression and different Blumeria graminis f. sp hordei resistance pattern.
The goal of the project is to obtain the GER4 locus sequence of those varieties.
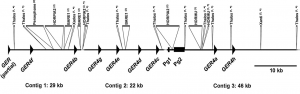
Barley genome is around 5.1 Gb and is mainly composed by repeated sequences (>80 %). In front of this complexity, building and screening non-gridded BAC library was the best approach to obtain this region with a high sequence quality that is necessary to make sequence comparison.
At the CNRGV, we are building and screening the 8 BAC libraries in order to identify the GER4 locus that is around 100kb long in size.
|
|
|

CNRGV's responsible
William Marande
Identification and sequencing of the asparagine synthetase genes in Durum Wheat
Identification and sequencing of the asparagine synthetase genes in Durum Wheat
Voir plus
Project coordinator :
Dr. Gabriella Sonnante
Institute of Plant Genetics (IGV) – CNR
Via Amendola, 165/A
70126 Bari (Italy)
Abstract :
It is well known that many plants rely on the synthesis and degradation of the amides asparagine and glutamine to relocate nitrogen within their tissue. In this context, asparagine plays a prominent role in nitrogen transport and storage in plants.
Glutamine-dependent asparagine synthetase (AsnS) catalyses the transfer of an amide group from glutamine to aspartate forming asparagine in an ATP-dependent reaction. In most plant species, AsnS seems to be encoded by a small gene family. The number of AsnS genes identified in plant species varies between one and four (i.e. one in asparagus, two in bread wheat and barley, three in Arabidopsis and sunflower, four in maize), and in most species only one or two genes are known.
The aim of this work is to identify and characterize AsnS genes in durum wheat. A hybridization of durum wheat macroarrays, representing a durum wheat BAC library accession, was carried out in order to find clones containing asparagine synthetase genes. Probes are developed on full-length AsnS sequences belonging to other close Triticeae taxa already present in NCBI. Positive BAC clones to hybridation will be validated by PCR and Sanger sequencing and entire BAC will be sequenced using 454 technology in order to obtain the different gene family members.
CNRGV involvement :
Responsible: William Marande

SUNRISE : SUNflower Resources to Improve yield Stability in a changing Environment
Project funded in the frame of "Investments for the future"
Voir plus
The world oilseed production will be faced to an increasing demand in the next thirty years catapulted by a combination of factors, including: i) an additional demand of edible oil, ii) the development of the biofuels industry and more specifically biodiesel around the world, and iii) the needs for green chemistry.
Sunflower represents a major renewable resource for food (oil), feed (meal), and green energy. France is a major sunflower producer. French breeders are ranked first in terms of sunflower seed production. World leading seed companies and SME involved in sunflower breeding are based in France and are creating a wide range of employments in France – from molecular breeders to farmers involved in seed production - , while ensuring a continuous genetic progress in integrating new technologies in their breeding programs.
During the past ten years, the impact of genetic advance on sunflower yield increase has been lower than expected, suggesting that current breeding resources and methods might not bring future solutions to the requested need in a context of climate changes. In order to face the challenges of delivering safe and high-quality food while maintaining yield and stability across different environments affected by climatic change, a paradigm shift is needed in sunflower breeding.
The near availability of the genome sequences of Helianthus annuus together with the breakthrough created by the new sequencing technologies, the development of phenotypic platforms and of bioinformatic tools allowing the integration of such high throughput data, are offering a favorable context to reinforce, through an optimization of the breeding process of hybrid cultivars, the competitiveness of French seed industry.
In an unprecedented effort (8 years project, 10 public and 7 private partners), SUNRISE offers unique opportunities to accelerate genetic gain and improve oil yield of sunflower hybrids grown under limited water supply through a better use of the outstanding genetic diversity of wild and domesticated Helianthus annuus genetic resources.
As the result of climate changes, more variability is expected in the timing and quantity of water availability for crop production from location to location. Sunflower is seen as a water stress tolerant crop, but wastes available water. On the long term period, due to the cost of investments all along the oil industry chain, the competitiveness of the sunflower chain is highly depending on the stability of oil yield across years and locations. SUNRISE will decipher the genes and genes networks involved in both the sunflower oil yield potential and its stability across years and locations, and then identify in the wide Helianthus annuus gene pool which alleles would be of interest for sunflower breeding.
Moreover, SUNRISE will devote a particular effort in the improvement of sunflower hybrid breeding process. SUNRISE will identify the loci and/or the heterosis mechanisms which are the most involved in homeostasis, depending on the parental alleles, and then build new gene pools exhibiting between themselves the better specific combining ability for homeostasis, i.e. yield stability.
All together, these two objectives will result in the definition of a new sunflower ideotype.
Full benefit of present possibilities requires (i) the better characterization of genetic diversity in order to optimize dense genotyping needed for genome wide association and linkage mapping, (ii) the development of appropriate detailed or high throughput phenotyping strategies, including molecular phenotyping, to characterize the sunflower response to the variation of the abiotic environment, (iii) the involvement of appropriate genetic design to decipher the genetic factors involved in this response, (iv) the integration of the knowledge into a crop model for in silico testing of the genotype environment interactions and for ideotypes design, (v) the development of innovative tools and methods to optimize the utilization of genetic resources and breed for new improved sunflower varieties for the concerned traits.
It relies on a large partnership between the key players of sunflower economy in France and in particular a strong private partnership unique to date in the history of sunflower research in France. This partnership will ensure that the new knowledge, resources and methods resulting from the project will be translated into products and varieties supporting the sunflower economy in France.
SUNRISE will ultimately enable efficient molecular based improvement to reduce the time from trait to commercialization. Indeed, SUNRISE will not only produce new resources, data and material, but will also follow their valorization by partners (usually competitors) that agree to synergize their efforts within the partnership of this project and give a feedback of their value in their programs. The project also integrates socio-economic analyses that will enable to evaluate the benefit of innovative genetic and ecophysiological results for the breeding sector and transfer of knowledge to agriculture.
Project coordinator:
Patrick VINCOURT
INRA UMR INRA-CNRS 441-2559
Chemin de Borde Rouge
31326 Castanet Tolosan
Patrick.Vincourt@toulouse.inra.fr
Project partner:
10 public and 7 private partners (CETIOM, BIOGEMMA,CAUSSADE SEMENCES, MAISADOUR, RAGT, SOLTIS,SYNGENTA SEEDS)
CNRGV involvement:
Responsible:
Arnaud Bellec
William Marande
Sonia Vautrin
Production and analysis of new genomic resources
![]()

Identification of the genomic region responsible for the resistance to the leaf rust in the weat variety Sinvalocho MA using non gridded BAC library.
Identification of the genomic region responsible for the resistance to the leaf rust in the weat variety Sinvalocho MA using non gridded BAC library.
Voir plus
Project coordinator:
Maria-José Dieguez
Instituto de Genética "Ewald A. Favret"
CICVyA, CNIA, INTA Castelar
C.C. 25 - Castelar (B1712WAA)
Buenos Aires – Argentina
Phone : 54 011 4450-0805/1876 int. 112
mdieguez@cnia.inta.gov.ar
Abstract:
The gene LrSV2 for adult plant wheat leaf rust resistance was mapped on sub-distal chromosome 3BS in the wheat cultivar Sinvalocho MA (Ingala et al, 2012). In a fine map of this region, close flanking markers were identified that were subsequently used to screen for recombinants within this target interval. The genotyping of these recombinants with additional markers allowed the narrowing of the LrSV2-containing interval to a distance that corresponds to 262 Kb in the variety Chinese Spring, whose sequence information is available.
The objective of this research was the construction of a non gridded BAC library of the wheat cultivar Sinvalocho MA and the identification of BAC clones spanning the LrSV2-containing interval.
Positive BAC clones that contain the region of interest will be sequenced using the 454 technology.
CNRGV involvement:
Responsible: William Marande

Evolutionary dynamics and genetic basis of dominance: sporophytic self-incompatibility in the Brassicaceae as a case study.
Evolutionary dynamics and genetic basis of dominance: sporophytic self-incompatibility in the Brassicaceae as a case study.
Voir plus
Project coordinator:
Vincent Castric
Unité Evo-Eco-Paléo (EEP) - UMR 8198
CNRS / Université de Lille - Sciences et Technologies
Batiment SN2, bureau 207
59655 Villeneuve d'Ascq - FRANCE
Project partners:
Hélène Touzet at the LIFL - Laboratoire d’Informatique Fondamentale de Lille
CNRS-INRIA- FRANCE
Isabelle Loisy and Thierry Gaude at the laboratory « Reproduction et Developpement des Plantes »
ENS Lyon CNRS - FRANCE
Website:
http://www.agence-nationale-recherche.fr/Project-ANR-11-JSV7-0008
http://eep.univ-lille.fr/fr/perso-vincent-castric
Abstract:
Dominance, the genetic phenomenon whereby one of the two alleles at a diploid locus is masked at the phenotypic level, is one of the earliest observations of classical genetics, yet also one whose genetic basis and evolution remain poorly understood. Dominance modifiers, i.e. genetic elements controlling the dominance of alleles at other genes, have been postulated by Ronald A. Fisher as early as 1928, but they were never documented since then. The question of whether these genetic elements indeed existed and how strongly natural selection could act on them violently opposed him to Sewall Wright in the 30’s.Earlier last year, Tarutani et al. (2010) identified, embedded within the gene cluster controlling self-incompatibility in the Brassicaceae, a trans-acting small RNA acting as dominance modifier of the gene controlling pollen specificity. This discovery provides an elegant mechanistic model for dominance/recessivity interaction in pollen, and hence the first description of dominance modifiers whose existence had been postulated since Fisher,but never documented yet. This opens up the exciting opportunity to address long-standing mechanistic as well as evolutionary issues, such as the type of mutations causing changes in dominance or how intensely natural selection acts on dominance modifiers. In this proposal, we propose to take advantage of this recent discovery and of our previous theoretical and empirical work to shed new light on the evolutionary dynamics and genetic basis of dominance, using an integrative multidisciplinary framework combining (1) phenotypic approaches to describe the dominance network among alleles at the gene controlling pollen self-incompatibility specificity in the plant Arabidopsis halleri; (2) genomic approaches to compare in silico the small RNA repertoire of dominant and recessive haplotypes and validate the proposed mechanism for the control of dominance in a genetic system characterized by strong multiallelism; (3) functional approaches to validate some of the candidate motifs; (4) theoretical approaches to explore how the control of dominance by small RNAs changes our understanding of how dominance can evolve in this genetic system, in particular focusing on the coevolution and asymetry it entails between small RNAs and their targets; (5) molecular evolution approaches to evaluate the level of functional constraint on small RNAs and their target sites.
CNRGV involvement:
BAC library construction and screening
Responsible: William Marande
Publications related to the project:
Evolutionary dynamics of genes controlling meiotic recombination in polyploid crop plants (DUPLIC)
Evolutionary dynamics of genes controlling meiotic recombination in polyploid crop plants (DUPLIC)
Voir plus
Meiotic recombination is a fundamental process for all sexual eukaryotes, which is required to produce balanced gametes and therefore contribute to the fertility and fitness of species. Meiotic recombination is also crucial for plant breeding because it allows, through the crossovers (COs), to reshuffle genetic material between individuals and between species. Huge international efforts have thus been made to identify the genes that are responsible for meiotic recombination in plants, using Arabidopsis thaliana, and to a lesser extent rice and maize, as model systems.
However our understanding still remains incomplete, in particular because the effect of polyploidy, which is a hallmark of plant genome evolution, has not been evaluated. We don't know the extent to which increased genome complexity in polyploid species is paralleled by an increased complexity in the genetic architecture underpinning crossover recombination. Are genes contributing to meiotic recombination randomly distributed between singleton and duplicated genes or conversely are they preferentially retained or preferentially lost after one episode of polyploidy? Are all the copies retained from the progenitor species expressed? Are they still functionally redundant? Or have they diversified following WGD? All these questions are still open and the very limited information available on these issues can not be integrated in a consistent way.
The DUPLIC project aims to advance understanding on the evolution of genes involved in meiotic recombination following polyploidy events. We will notably try to decipher if preferential elimination of duplicated copies is a general, thus predictable response in polyploid species or conversely if polyploidy may foster genetic innovation in the control of CO formation.
One on the research objective will be to determine the patterns of duplicated copy retention/loss for 18 recombination genes in two complementary polyploid crop species, oilseed rape (Brassica napus) and bread wheat (Triticum aestivum).
Project coordinator
Project Partners
CNRGV's Responsible
CNRGV involvement:
Isolation, sequencing and characterization of the various copies of the selected genes in both oilseed rape and wheat through PCR or hybridization of the of oilseed rape and wheat BAC libraries
Funded by ANR (The French National Research Agency): ANR DUPLIC
|
|
|


Publication related to the project


A reference genome assembly for Quercus canariensis Willd
Authors
F. Couturier, C. Cravero, I. Lesur, J. Confais, E. Belmonte, L. Piat, W. Marande, C. Rellstab, M. Valbuena, E. Saez-Laguna, L. Duvaux
doi:
Voir plus
Abstract
We present a genome assembly from a specimen of Quercus canariensis (Fagaceae; Fagales; Magnoliopsida). The assembly was generated using PacBio HiFi long reads with an approximate sequencing depth of 39X and scaffolded using a reference-guided approach. The genome sequence has a total length of 816.0 megabases for haplotype 1 and 804.8 megabases for haplotype 2. The two haplotypes are each resolved into 12 chromosomal pseudomolecules, with only 3.48% and 1.36% of sequences remaining unplaced in haplotypes 1 and 2, respectively. Assembly completeness is supported by BUSCO scores of 98.3% and 98.2% complete genes for haplotypes 1 and 2, respectively. Structural annotation identified 51,882 and 46,482 protein-coding genes in haplotypes 1 and 2, respectively. This genome assembly provides the first chromosome-scale reference genome for Q. canariensis, laying the base for future genomic and evolutionary studies in this understudied species of the hybridizing white oak species complex.
Taxonomy Lineage cellular organisms; Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta; Spermatophyta; Magnoliopsida; eudicotyledons; Gunneridae; Pentapetalae; rosids; fabids; Fagales; Fagaceae; Quercus
EBI:txid568684
Quercus canariensis Willd. 1809 (Willdenow)







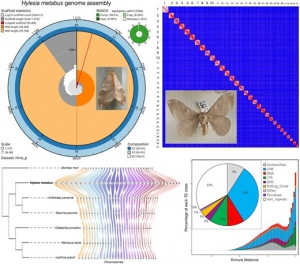





Cantaloupe melon genome reveals 3D chromatin features and structural relationship with the ancestral Cucurbitaceae karyotype.
Clement Pichot, Anis Djari, Joseph Tran, Marion Verdenaud, William Marande, Cecile Huneau, Veronique Gautier, David Latrasse, Sandrine Arribat, Vivien Sommard, Christelle Troadec, Charles Poncet, Mohammed Bendahmane, Judit Szecsi, Catherine Dogimont, Jerome Salse, Moussa Benhamed, Mohamed Zouine, Adnane Boualem, Abdelhafid Bendahmane.
Voir plus
Journal: IScience
https://doi.org/10.1016/j.isci.2021.1036
Abstract
Cucumis melo displays a large diversity of horticultural groups with Cantaloupe melon the most cultivated type. Using a combination of single-molecule sequencing, 10X Genomics link-reads, high-density optical and genetic maps and chromosome conformation capture (Hi-C) we assembled a chromosome scale C.melo var. cantalupensis Charentais mono genome. Integration of RNA-seq, MeDip-seq, ChIP-seq and Hi-C data revealed a widespread compartmentalization of the melon genome, segregating constitutive heterochromatin and euchromatin. Genome-wide comparative and evolutionary analysis between melon botanical groups identified Charentais mono genome increasingly more divergent from Harukei-3 (reticulatus), Payzawat (inodorus), and HS (ssp. agrestis) genomes. To assess the paleohistory of the Cucurbitaceae, we reconstructed the ancestral Cucurbitaceae karyotype and compared it to sequenced cucurbit genomes. In contrast to other species that experienced massive chromosome shuffling, melon has retained the ancestral genome structure. We provide comprehensive genomic resources and new insights in the diversity of melon horticultural groups and evolution of cucurbits.






A wheat cysteine-rich receptor-like kinase confers broad-spectrum resistance against Septoria tritici blotch
Cyrille Saintenac, Florence Cambon, Lamia Aouini, Els Verstappen, Seyed Mahmoud Tabib Ghaffary, Théo Poucet, William Marande, Hélène Berges, Steven Xu, Maëlle Jaouannet, Bruno Favery, Julien Alassimone, Andrea Sánchez-Vallet, Justin Faris, Gert Kema, Oliver Robert & Thierry Langin
Voir plus
The poverty of disease resistance gene reservoirs limits the breeding of crops for durable resistance against evolutionary dynamic pathogens. Zymoseptoria tritici which causes Septoria tritici blotch (STB), represents one of the most genetically diverse and devastating wheat pathogens worldwide. No fully virulent Z. tritici isolates against synthetic wheats carrying the major resistant gene Stb16q have been identified. Here, we use comparative genomics, mutagenesis and complementation to identify Stb16q, which confers broad-spectrum resistance against Z. tritici. The Stb16q gene encodes a plasma membrane cysteine-rich receptor-like kinase that was recently introduced into cultivated wheat and which considerably slows penetration and intercellular growth of the pathogen.



The unusual S locus of Leavenworthia is composed of two sets of paralogous loci
Chantha SC, Herman AC, Castric V, Vekemans X, Marande W, Schoen DJ.
Journal: New Phytologist
DOI: 10.1111/nph.14764
Voir plus
Chantha SC(1), Herman AC(1,2), Castric V(3), Vekemans X(3), Marande W(4), Schoen DJ(1).
Author information
(1) Department of Biology, McGill University, 1205 Avenue Docteur Penfield, Montreal, QC, Canada,H3A1B1.
(2) Department of Plant Biology, University of Minnesota, St Paul, MN, 55108, USA.
(3) Unité Evo-Eco-Paléo (EEP) - UMR 8198, CNRS/Université de Lille - Sciences et Technologies, Villeneuve d'Ascq Cedex, F-59655, France
(4) Institut National de la Recherche Agronomique, 31326, Castanet Tolosan Cedex, France.
Journal: New Phytologist
DOI: 10.1111/nph.14764
Abstract
The Leavenworthia self-incompatibility locus (S locus) consists of paralogs (Lal2, SCRL) of the canonical Brassicaceae S locus genes (SRK, SCR), and is situated in a genomic position that differs from the ancestral one in the Brassicaceae. Unexpectedly, in a small number of Leavenworthia alabamica plants examined, sequences closely resembling exon 1 of SRK have been found, but the function of these has remained unclear. BAC cloning and expression analyses were employed to characterize these SRK-like sequences. An SRK-positive Bacterial Artificial Chromosome clone was found to contain complete SRK and SCR sequences located close by one another in the derived genomic position of the Leavenworthia S locus, and in place of the more typical Lal2 and SCRL sequences. These sequences are expressed in stigmas and anthers, respectively, and crossing data show that the SRK/SCR haplotype is functional in self-incompatibility. Population surveys indicate that < 5% of Leavenworthia S loci possess such alleles. An ancestral translocation or recombination event involving SRK/SCR and Lal2/SCRL likely occurred, together with neofunctionalization of Lal2/SCRL, and both haplotype groups now function as Leavenworthia S locus alleles. These findings suggest that S locus alleles can have distinctly different evolutionary origins.
Link: http://onlinelibrary.wiley.com/doi/10.1111/nph.14764/full
Genome sequencing reveals the origin of the allotetraploid arabidopsis suecica
Novikova PY, Tsuchimatsu T, Simon S, Nizhynska V, Voronin V, Burns R, Fedorenko OM, Holm S, Saell T, Prat E, Marande W, Castric V, Nordborg M.
Journal: Molecular Biology and Evolution
DOI: 34: 957-968.
Voir plus
Novikova PY, Tsuchimatsu T, Simon S, Nizhynska V, Voronin V, Burns R, Fedorenko OM, Holm S, Saell T, Prat E, Marande W, Castric V, Nordborg M.
Journal: Molecular Biology and Evolution
DOI: 34: 957-968.
Abstract
Polyploidy is an example of instantaneous speciation when it involves the formation of a new cytotype that is incompatible with the parental species. Because new polyploid individuals are likely to be rare, establishment of a new species is unlikely unless polyploids are able to reproduce through self-fertilization (selfing), or asexually. Conversely, selfing (or asexuality) makes it possible for polyploid species to originate from a single individual—a bona fide speciation event. The extent to which this happens is not known. Here, we consider the origin of Arabidopsis suecica, a selfing allopolyploid between Arabidopsis thaliana and Arabidopsis arenosa, which has hitherto been considered to be an example of a unique origin. Based on whole-genome re-sequencing of 15 natural A. suecica accessions, we identify ubiquitous shared polymorphism with the parental species, and hence conclusively reject a unique origin in favor of multiple founding individuals. We further estimate that the species originated after the last glacial maximum in Eastern Europe or central Eurasia (rather than Sweden, as the name might suggest). Finally, annotation of the self-incompatibility loci in A. suecica revealed that both loci carry non-functional alleles. The locus inherited from the selfing A. thaliana is fixed for an ancestral non-functional allele, whereas the locus inherited from the outcrossing A. arenosa is fixed for a novel loss-of-function allele. Furthermore, the allele inherited from A. thaliana is predicted to transcriptionally silence the allele inherited from A. arenosa, suggesting that loss of self-incompatibility may have been instantaneous.
Patterns of polymorphism at the self-incompatibility locus in 1,083 Arabidopsis thaliana genomes.
Mol Biol Evol. doi: 10.1093/molbev/msx122.
Voir plus
Authors :
Tsuchimatsu T, Goubet PM, Gallina S, Holl AC, Fobis-Loisy I, Bergès H, Marande W, Prat E, Meng D, Long Q, Platzer A, Nordborg M, Vekemans X, Castric V.
Mol Biol Evol. doi: 10.1093/molbev/msx122.
Abstract :
Although the transition to selfing in the model plant Arabidopsis thaliana involved the loss of the self-incompatibility (SI) system, it clearly did not occur due to the fixation of a single inactivating mutation at the locus determining the specificities of SI (the S-locus). At least three groups of divergent haplotypes (haplogroups), corresponding to ancient functional S-alleles, have been maintained at this locus, and extensive functional studies have shown that all three carry distinct inactivating mutations. However, the historical process of loss of SI is not well understood, in particular its relation with the last glaciation. Here, we took advantage of recently published genomic re-sequencing data in 1,083 Arabidopsis thaliana accessions that we combined with BAC sequencing to obtain polymorphism information for the whole S-locus region at a species-wide scale. The accessions differed by several major rearrangements including large deletions and inter-haplogroup recombinations, forming a set of haplogroups that are widely distributed throughout the native range and largely overlap geographically. 'Relict' A. thaliana accessions that directly derive from glacial refugia are polymorphic at the S-locus, suggesting that the three haplogroups were already present when glacial refugia from the last Ice Age became isolated. Inter-haplogroup recombinant haplotypes were highly frequent, and detailed analysis of recombination breakpoints suggested multiple independent origins. These findings suggest that the complete loss of SI in A. thaliana involved independent self-compatible mutants that arose prior to the last Ice Age, and experienced further rearrangements during post-glacial colonization.
Extraction of high-molecular-weight genomic DNA for long-read sequencing of single molecules
Biotechniques. 2016 Oct 1;61(4):203-205.
Voir plus
Authors
Mayjonade B, Gouzy J, Donnadieu C, Pouilly N, Marande W, Callot C, Langlade N, Muños S.
Biotechniques. 2016 Oct 1;61(4):203-205.
Abstract
De novo sequencing of complex genomes is one of the main challenges for researchers seeking high-quality reference sequences. Many de novo assemblies are based on short reads, producing fragmented genome sequences. Third-generation sequencing, with read lengths >10 kb, will improve the assembly of complex genomes, but these techniques require high-molecular-weight genomic DNA (gDNA), and gDNA extraction protocols used for obtaining smaller fragments for short-read sequencing are not suitable for this purpose. Methods of preparing gDNA for bacterial artificial chromosome (BAC) libraries could be adapted, but these approaches are time-consuming, and commercial kits for these methods are expensive. Here, we present a protocol for rapid, inexpensive extraction of high-molecular-weight gDNA from bacteria, plants, and animals. Our technique was validated using sunflower leaf samples, producing a mean read length of 12.6 kb and a maximum read length of 80 kb.
Comparative Analysis of WRKY Genes Potentially Involved in Salt Stress Responses in Triticum turgidum L. ssp. durum.
Front Plant Sci 7:2034. doi: 10.3389/fpls.2016.02034
Voir plus
Authors :
Yousfi FE, Makhloufi E, Marande W, Ghorbel AW., Bouzayen M and Bergès H
Front Plant Sci 7:2034. doi: 10.3389/fpls.2016.02034
Abstract :
WRKY transcription factors are involved in multiple aspects of plant growth, development and responses to biotic stresses. Although they have been found to play roles in regulating plant responses to environmental stresses, these roles still need to be explored, especially those pertaining to crops. Durum wheat is the second most widely produced cereal in the world. Complex, large and unsequenced genomes, in addition to a lack of genomic resources, hinder the molecular characterization of tolerance mechanisms. This paper describes the isolation and characterization of five TdWRKY genes from durum wheat (Triticum turgidum L. ssp. durum). A PCR-based screening of a T. turgidum BAC genomic library using primers within the conserved region of WRKY genes resulted in the isolation of five BAC clones. Following sequencing fully the five BACs, fine annotation through Triannot pipeline revealed 74.6% of the entire sequences as transposable elements and a 3.2% gene content with genes organized as islands within oceans of TEs. Each BAC clone harbored a TdWRKY gene. The study showed a very extensive conservation of genomic structure between TdWRKYs and their orthologs from Brachypodium, barley, and T. aestivum. The structural features of TdWRKY proteins suggested that they are novel members of the WRKY family in durum wheat. TdWRKY1/2/4, TdWRKY3, and TdWRKY5 belong to the group Ia, IIa, and IIc, respectively. Enrichment of cis-regulatory elements related to stress responses in the promoters of some TdWRKY genes indicated their potential roles in mediating plant responses to a wide variety of environmental stresses. TdWRKY genes displayed different expression patterns in response to salt stress that distinguishes two durum wheat genotypes with contrasting salt stress tolerance phenotypes. TdWRKY genes tended to react earlier with a down-regulation in sensitive genotype leaves and with an up-regulation in tolerant genotype leaves. The TdWRKY transcripts levels in roots increased in tolerant genotype compared to sensitive genotype. The present results indicate that these genes might play some functional role in the salt tolerance in durum wheat.
Long Read Sequencing Technology to Solve Complex Genomic Regions Assembly in Plants.
Journal of Next Generation Sequencing & Applications.
Voir plus
Authors :
Arnaud Bellec, Audrey Courtial, Stephane Cauet, Nathalie Rodde, Sonia Vautrin, Genseric Beydon, Nadege Arnal, Nadine Gautier, Joelle Fourment, Elisa Prat, William Marande, Yves Barriere and Helene Berges.
Journal of Next Generation Sequencing & Applications.
Abstract :
Background:
Numerous completed or on-going whole genome sequencing projects have highlighted the fact that obtaining a high quality genome sequence is necessary to address comparative genomics questions such as structural variations among genotypes and gain or loss of specific function. Despite the spectacular progress that has been made in sequencing technologies, obtaining accurate and reliable data is still a challenge, both at the whole genome scale and when targeting specific genomic regions. These problems are even more noticeable for complex plant genomes. Most plant genomes are known to be particularly challenging due to their size, high density of repetitive elements and various levels of ploidy. To overcome these problems, we have developed a strategy to reduce genome complexity by using the large insert BAC libraries combined with next generation sequencing technologies.
Results:
We compared two different technologies (Roche-454 and Pacific Biosciences PacBio RS II) to sequence pools of BAC clones in order to obtain the best quality sequence. We targeted nine BAC clones from different species (maize, wheat, strawberry, barley, sugarcane and sunflower) known to be complex in terms of sequence assembly. We sequenced the pools of the nine BAC clones with both technologies. We compared assembly results and highlighted differences due to the sequencing technologies used.
Conclusions:
We demonstrated that the long reads obtained with the PacBio RS II technology serve to obtain a better and more reliable assembly, notably by preventing errors due to duplicated or repetitive sequences in the same region.
Link :
Dominance hierarchy arising from the evolution of a complex small RNA regulatory network.
Science. 346(6214):1200-5. doi: 10.1126/science.1259442.
Voir plus
Authors
Durand E1, Méheust R1, Soucaze M1, Goubet PM1, Gallina S1, Poux C1, Fobis-Loisy I2, Guillon E2, Gaude T2, Sarazin A3, Figeac M4, Prat E5, Marande W5, Bergès H5, Vekemans X1, Billiard S1, Castric V6.
Science. 346(6214):1200-5. doi: 10.1126/science.1259442.
Abstract
The prevention of fertilization through self-pollination (or pollination by a close relative) in the Brassicaceae plant family is determined by the genotype of the plant at the self-incompatibility locus (S locus). The many alleles at this locus exhibit a dominance hierarchy that determines which of the two allelic specificities of a heterozygous genotype is expressed at the phenotypic level. Here, we uncover the evolution of how at least 17 small RNA (sRNA)-producing loci and their multiple target sites collectively control the dominance hierarchy among alleles within the gene controlling the pollen S-locus phenotype in a self-incompatible Arabidopsis species. Selection has created a dynamic repertoire of sRNA-target interactions by jointly acting on sRNA genes and their target sites, which has resulted in a complex system of regulation among alleles.
Isolation and molecular characterization of ERF1, an ethylene response factor gene from durum wheat (Triticum turgidum L. subsp. durum), potentially involved in salt-stress responses.
J Exp Bot. 2014
Voir plus
Authors
Makhloufi E, Yousfi FE, Marande W, Mila I, Hanana M, Bergès H, Mzid R, Bouzayen M.
J Exp Bot. 2014
Abstract
As food crop, wheat is of prime importance for human society. Nevertheless, our understanding of the genetic and molecular mechanisms controlling wheat productivity conditions has been, so far, hampered by the lack of sufficient genomic resources. The present work describes the isolation and characterization of TdERF1, an ERF gene from durum wheat (Triticum turgidum L. subsp. durum). The structural features of TdERF1 supported the hypothesis that it is a novel member of the ERF family in durum wheat and, considering its close similarity to TaERF1 of Triticum aestivum, it probably plays a similar role in mediating responses to environmental stresses. TdERF1 displayed an expression pattern that discriminated between two durum wheat genotypes contrasted with regard to salt-stress tolerance. The high number of cis-regulatory elements related to stress responses present in the TdERF1 promoter and the ability of TdERF1 to regulate the transcription of ethylene and drought-responsive promoters clearly indicated its potential role in mediating plant responses to a wide variety of environmental constrains. TdERF1 was also regulated by abscisic acid, ethylene, auxin, and salicylic acid, suggesting that it may be at the crossroads of multiple hormone signalling pathways. Four TdERF1 allelic variants have been identified in durum wheat genome, all shown to be transcriptionally active. Interestingly, the expression of one allelic form is specific to the tolerant genotype, further supporting the hypothesis that this gene is probably associated with the susceptibility/tolerance mechanism to salt stress. In this regard, the TdERF1 gene may provide a discriminating marker between tolerant and sensitive wheat varieties.
Meiotic gene evolution: can you teach a new dog new tricks?
Mol Biol Evol. 2014
Voir plus
Authors
Lloyd A, Ranoux M, Vautrin S, Glover N, Fourment J, Charif D, Choulet F, Lassalle G, Marande W, Tran J, Granier F, Pingault L, Remay A, Marquis C, Belcram H, Chalhoub B, Feuillet C, Bergès H, Sourdille P, Jenczewski E.
MolBiol Evol. 2014
Abstract
Meiosis, the basis of sex, evolved through iterative gene duplications. To understand whether subsequent duplications have further enriched the core meiotic "tool-kit", we investigated the fate of meiotic gene duplicates following Whole Genome Duplication (WGD), a common occurrence in eukaryotes. We show that meiotic genes return to a single copy more rapidly than genome-wide average in Angiosperms, one of the lineages in which WGD is most vividly exemplified. The rate at which duplicates are lost decreases through time, a tendency that is also observed genome-wide and may thus prove to be a general trend post-WGD. The sharpest decline is observed for the subset of genes mediating meiotic recombination; however, we found no evidence that the presence of these duplicates is counter-selected in two recent polyploid crops selected for fertility. We therefore propose that their loss is passive, highlighting how quickly WGDs are resolved in the absence of selective duplicate retention.