

- Badouin H, et al., (2017) The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature. 546 (7656):148-152. doi: 10.1038/nature22380.
- Kane, N.C., Gill, N., King, M.G., Bowers, J.E., Bergès, H., Gouzy, J., Bachlava, E., Langlade, N.B., Lai, Z., Stewart, M., Burke, J.M., Vincourt, P., Knapp, S.J., and Rieseberg L.H. (2011) Progress towards a reference genome for sunflower. Botany (89:429-437)
- Construction and screening of a Non Gridded BAC library
- Physical map of the genomic region of interest
- Sonia Vautrin
- Construction et Criblage d’une banque BAC de panais
- Séquençage
- Arnaud Bellec
- 3D pool production and screening
- BAC libraires construction
- Sequence-Based Analysis of Structural Organization and Composition of the Cultivated Sunflower (Helianthus annuus L.) Genome
- Progress towards a reference genome for sunflower
- Sonia Vautrin
- A mosaic monoploid reference sequence for the highly complex genome of sugarcane
- Building the sugarcane genome for biotechnology and identifying evolutionary trends
- The Sugarcane Genome Challenge: Strategies for Sequencing a Highly Complex Genome
- Sonia Vautrin
-
Italian National Agency for New technologies, Energy and Environment (ENEA).
ENEA-Casaccia RC, BAS BIOTEC-GEN
Via Anguillarese, 301
00123 Rome
Italy -
Dr. Monder Bouzayen
Laboratoire de Génomique et Biotechnologies des fruits
UMR990
Chemin de Borde Rouge
31326 Castanet Tolosan Dr. Catherine Feuillet
Institut National de la Recherche Agronomique-ASP
Domaine de Crouelle
234 Avenue du Brezet
63100 Clermont-Ferrand
France
www.clermont.inra.fr/umr-aspE-mail : catherine.feuillet@clermont.inra.fr
- La construction et l’ancrage des cartes physiques du blé et de l'orge pour les chromosomes 1 et 3 qui portent un grand nombre de caractères agronomiques importants (par ex. la résistance de maladie, la production et la qualité)
- L’isolement de gènes et QTLs liés à la résistance à des maladies, le rendement et les caractères de qualité pour le blé et l'orge
- L’identification et l’exploitation de nouveaux allèles pour les gènes isolés par l'utilisation de variants naturels et de populations mutées aussi bien que le germplasm sauvage
- Le développement de nouvelles variétés pour les besoins des consommateurs et des agriculteurs à travers des approches moléculaires
- Le développement de nouveaux outils bioinformatiques pour structurer, centraliser et analyser à grande échelle les données génomiques qui seront générées dans ce projet
- La mise en place de formation pour ces nouvelles approches technologiques, le partage des résultats et les transferts technologiques à l'industrie et aux collaborateurs internationaux.
- Institut National de la Recherche Agronomique - INRA - France
- Leibniz Institute of Plant Genetics and Crop Plant Research - IPK - Germany
- Institute of Experimental Botany - IEB - Czech Republic
- GSF- National Research Center for Environment and Health - GSF - Germany
- Università degli Studi di Milano - UMIL - Italy
- University of Haifa - HU - Israel
- MTT (Maa- ja elintarviketalouden tutkimuskeskus) - MTT - Finland
- Scottish Crop Research Institute - SCRI - UK
- Sabanci University - SABA - Turkey
- National Institute of Agricultural Botany - NIAB - UK
- John Innes Centre - JIC - UK
- Universität Zürich - UZH - Switzerland
- INRA Transfert - IT - France
- Biogemma - BGA - France
- Lochow-Petkus GmbH - LP - Germany
- Istituto di Genomica Applicata - IGA - Italy
- University of Bologna -UNBO - Italy
Dr. Patrick Vincourt
LIPM (UMR 441-2594 INRA-CNRS)
BP 52627
Chemin de Borde Rouge - Auzeville
31326 Castanet Tolosan
Email : patrick.vincourt@toulouse.inra.frLaboratoire “ Interactions Plantes Microorganismes”, UMR 441-2594(INRA-CNRS)
INRA-CNRS
31326 CASTANET TOLOSANDirecteur du laboratoire : Pascal GAMAS
Responsable scientifique : Patrick VINCOURTUMR 1095 “ Amélioration et Santé des Plantes ”
INRA – Université Blaise Pascal
63100 CLERMONT-FERRANDDirecteur du laboratoire : Gilles CHARMET
Responsable scientifique : Felicity VEAR - D.TOURVIEILLEUMR 1065 « Santé végétale »des Plantes
INRA/ENITA Bordeaux
33883 VILLENAVE-D'ORNON CEDEXDirecteur du laboratoire : D. THIERRY
Responsable scientifique : F. DELMOTTEUR 1258 Centre National de Ressources Génomiques Végétales
CNRGV -INRA
31326 CASTANET TOLOSANDirecteur du laboratoire/Responsable scientifique : Helène BERGES
-
Unité de Génétique et Amélioration des Fruits et Légumes
INRA-GAFL, UR1052
84143 Montfavet Cedex
Directeur du laboratoire : Mathilde Causse
Responsable scientifique : Véronique Lefebvre -
Unité de Recherche en Génomique Végétale
URGV, UMR1165
INRA-CNRS-Univ
91057 Evry Cedex
Directeur du laboratoire : Michel Caboche
Responsable scientifique :Abdelhafifid Bendahmane -
Centre National de Ressources Génomiques Végétales
INRA-CNRGV, UR1258
31326 Castanet Tolosan Cedex
Directeur du laboratoire / Responsable scientifique : Hélène Bergès
International Consortium on Sunflower Genomics - ICSG
The domesticated sunflower, Helianthus annuus L., is a global oil crop that has promise for climate change adaptation, because it can maintain stable yields across a wide variety of environmental conditions, including drought. Even greater resilience is achievable through the mining of resistance alleles from compatible wild sunflower relatives, including numerous extremophile species. The consortium has established a high-quality reference for the sunflower genome (3.6 Gb).
Voir plus
The domesticated sunflower, Helianthus annuus L., is a global oil crop that has promise for climate change adaptation, because it can maintain stable yields across a wide variety of environmental conditions, including drought. Even greater resilience is achievable through the mining of resistance alleles from compatible wild sunflower relatives, including numerous extremophile species. The consortium has established a high-quality reference for the sunflower genome (3.6 Gb). The genome mostly consists of highly similar, related sequences and required single-molecule real-time sequencing technologies for successful assembly. Genome analyses enabled the reconstruction of the evolutionary history of the Asterids, further establishing the existence of a whole-genome triplication at the base of the Asterids II clade and a sunflower-specific whole-genome duplication around 29 million years ago. An integrative approach combining quantitative genetics, expression and diversity data permitted development of comprehensive gene networks for two major breeding traits, flowering time and oil metabolism, and revealed new candidate genes in these networks. The genomic architecture of flowering time has been shaped by the most recent whole-genome duplication, which suggests that ancient paralogues can remain in the same regulatory networks for dozens of millions of years. This genome represents a cornerstone for future research programs aiming to exploit genetic diversity to improve biotic and abiotic stress resistance and oil production, while also considering agricultural constraints and human nutritional needs.
Collaborators
Dr Stéphane Muños - Institut National de la Recherche Agronomique, “INRA”, France
Dr Loren RIESEBERG - University of British Columbia, Canada
Dr John BURKE - University of Georgia, USA
Dr Sariel HUBNER - Galilee Research Institute (MIGAL), Israel
Articles
CNRGV's responsible
Sonia Vautrin

Pea MUlti-STress adaptation and biological regulations for yield improvement and stability
Project funded in the frame of "Investments for the future"
Voir plus
Project coordinator:
Judith BURSTIN
INRA
UMR AgroEcologie
17 rue Sully
BP 86510
21065 DIJON
Email : judith.burstin@dijon.inra.fr
Website: https://www.peamust-project.fr/Le-projet
Project partner:
The PeaMUST project combines 26 public and private partners.
Abstract
• Develop novel pea varieties and optimize plant-symbiotic interactions for stabilizing seed yield and quality, in the context of pesticide reduction and climate change
• Enhance national and international research and competitiveness of the French breeders and pea production
Objectives
• Undertake a program of genomic selection targeted to low-input cropping systems
• Discover molecular determinants of disease, insect and frost partial resistance in pea and faba bean
• Investigate the potential of architecture and plant-symbiote interactions in breeding for multiple stress tolerance
• Provide enhanced platforms for gene validation in pea
• Devise best breeding strategies towards optimized yield and stress tolerance
Methodology
• Federate actors gathering various complementary competencies (quantitative genetics, breeding, genomics, biochemistry, biotechnology, plant /pathogen, pests and symbiots interactions, agronomy, socio-economy)
• Use associated phenotyping platforms (PPHD Dijon, DIAPHEN Montpellier, UE Epoisses, UE Le Rheu, UE Mons, UE Clermont-Theix, UE Lusignan, UE Toulouse, SNES) and national sequencing and genotyping platforms (GENOTOUL, GENTYANE, GENOSCOPE)
Main Results
• Acquire original basic knowledge on the genetic control of multiple stress tolerance (QTL cloning, impact of plant architecture, phenology and symbiotic interactions on multiple-stress tolerance)
• Develop unique tools for gene identification: high throughput sequence-based genotyping, translational genomic tools, association genetics panels, TILLING and VIGS platforms, user-friendly databases
• Develop enhanced breeding methods and lines, optimizing yield under varying stress conditions
CNRGV's responsibles:
Sonia Vautrin - Genséric Beydon
CNRGV involvement:
Bac libraries construction
![]()
Publication related to the project:
Development of a Sequence-Based Reference Physical Map of Pea (Pisum sativum L.).

Functional genomics of the wheat-Fusarium interaction
Functional genomics of the wheat-Fusarium interaction
Voir plus
|
Project coordinator:
Hermann BÜRSTMAYR / Wolfgang SCHWEIGER |
|

Abstract:
This project intends to fine-map one of the most effective Fusarium-resistance quantitative trail locus (QTL) Qfhs.ifa-5A in wheat. Qfhs.ifa-5A is located on the short arm of chromosome 5A supposedly close to the centromere. The region exhibits a very low recombination rate. To generate a high-resolution map for this region we will employ several strategies from screening for meiototic recombination events to physical mapping approaches that will provide additional breakpoints in this poor-recombining region. Using markers close to the physical position of the QTL, we will screen a non-gridded BAC library of the resistant wheat cultivar CM-82036. Selected BACs will be sequenced and investigated for more markers and candidate genes underlying the QTL.
CNRGV involvement:
CNRGV's responsible
Publication related to the project:

SUNRISE : SUNflower Resources to Improve yield Stability in a changing Environment
Project funded in the frame of "Investments for the future"
Voir plus
The world oilseed production will be faced to an increasing demand in the next thirty years catapulted by a combination of factors, including: i) an additional demand of edible oil, ii) the development of the biofuels industry and more specifically biodiesel around the world, and iii) the needs for green chemistry.
Sunflower represents a major renewable resource for food (oil), feed (meal), and green energy. France is a major sunflower producer. French breeders are ranked first in terms of sunflower seed production. World leading seed companies and SME involved in sunflower breeding are based in France and are creating a wide range of employments in France – from molecular breeders to farmers involved in seed production - , while ensuring a continuous genetic progress in integrating new technologies in their breeding programs.
During the past ten years, the impact of genetic advance on sunflower yield increase has been lower than expected, suggesting that current breeding resources and methods might not bring future solutions to the requested need in a context of climate changes. In order to face the challenges of delivering safe and high-quality food while maintaining yield and stability across different environments affected by climatic change, a paradigm shift is needed in sunflower breeding.
The near availability of the genome sequences of Helianthus annuus together with the breakthrough created by the new sequencing technologies, the development of phenotypic platforms and of bioinformatic tools allowing the integration of such high throughput data, are offering a favorable context to reinforce, through an optimization of the breeding process of hybrid cultivars, the competitiveness of French seed industry.
In an unprecedented effort (8 years project, 10 public and 7 private partners), SUNRISE offers unique opportunities to accelerate genetic gain and improve oil yield of sunflower hybrids grown under limited water supply through a better use of the outstanding genetic diversity of wild and domesticated Helianthus annuus genetic resources.
As the result of climate changes, more variability is expected in the timing and quantity of water availability for crop production from location to location. Sunflower is seen as a water stress tolerant crop, but wastes available water. On the long term period, due to the cost of investments all along the oil industry chain, the competitiveness of the sunflower chain is highly depending on the stability of oil yield across years and locations. SUNRISE will decipher the genes and genes networks involved in both the sunflower oil yield potential and its stability across years and locations, and then identify in the wide Helianthus annuus gene pool which alleles would be of interest for sunflower breeding.
Moreover, SUNRISE will devote a particular effort in the improvement of sunflower hybrid breeding process. SUNRISE will identify the loci and/or the heterosis mechanisms which are the most involved in homeostasis, depending on the parental alleles, and then build new gene pools exhibiting between themselves the better specific combining ability for homeostasis, i.e. yield stability.
All together, these two objectives will result in the definition of a new sunflower ideotype.
Full benefit of present possibilities requires (i) the better characterization of genetic diversity in order to optimize dense genotyping needed for genome wide association and linkage mapping, (ii) the development of appropriate detailed or high throughput phenotyping strategies, including molecular phenotyping, to characterize the sunflower response to the variation of the abiotic environment, (iii) the involvement of appropriate genetic design to decipher the genetic factors involved in this response, (iv) the integration of the knowledge into a crop model for in silico testing of the genotype environment interactions and for ideotypes design, (v) the development of innovative tools and methods to optimize the utilization of genetic resources and breed for new improved sunflower varieties for the concerned traits.
It relies on a large partnership between the key players of sunflower economy in France and in particular a strong private partnership unique to date in the history of sunflower research in France. This partnership will ensure that the new knowledge, resources and methods resulting from the project will be translated into products and varieties supporting the sunflower economy in France.
SUNRISE will ultimately enable efficient molecular based improvement to reduce the time from trait to commercialization. Indeed, SUNRISE will not only produce new resources, data and material, but will also follow their valorization by partners (usually competitors) that agree to synergize their efforts within the partnership of this project and give a feedback of their value in their programs. The project also integrates socio-economic analyses that will enable to evaluate the benefit of innovative genetic and ecophysiological results for the breeding sector and transfer of knowledge to agriculture.
Project coordinator:
Patrick VINCOURT
INRA UMR INRA-CNRS 441-2559
Chemin de Borde Rouge
31326 Castanet Tolosan
Patrick.Vincourt@toulouse.inra.fr
Project partner:
10 public and 7 private partners (CETIOM, BIOGEMMA,CAUSSADE SEMENCES, MAISADOUR, RAGT, SOLTIS,SYNGENTA SEEDS)
CNRGV involvement:
Responsible:
Arnaud Bellec
William Marande
Sonia Vautrin
Production and analysis of new genomic resources
![]()

Identification et caractérisation enzymatique des chainons moléculaires manquant dans la voie de biosynthèse des furocoumarines chez le panais (Pastinaca sativa)
Projet en collaboration avec le laboratoire Agronomie et Environnement de Vandoeuvre.
Voir plus
Coordinateur du projet :
Prof. Frederic Bourgaud
Directeur du laboratoire Agronomie et Environnement
Dr Sandro ROSELLI - Post-Doctorant
Partenaire :
Laboratoire Agronomie et Environnement
UMR 1121 Université de Lorraine – INRA
ENSAIA, 2 Avenue Forêt de Haye
F-54500 Vandoeuvre
Résumé :
Les furocoumarines sont des composés secondaires issus du métabolisme des phenylpropanoïdes et principalement distribués chez quatre familles de végétaux supérieurs : Apiacées (céleri), Moracées (ficus), Rutacées (citrus) et Fabacées (coronilles). Certaines de ces plantes, comme les Apiacées (carotte, céleri, persil) ou les Rutacées (Citrus), ont un intérêt agronomique évident. Chez ces espèces à usage alimentaire, les furocoumarines ont fait l’objet d’études car elles présentent un risque de toxicité majeur pour les consommateurs de ces produits végétaux. D’une manière plus générale, les plantes à furocoumarines présentent un intérêt fondamental dans le domaine de la biologie végétale dans la mesure où elles constituent un des modèles les mieux compris de co-évolution avec des insectes herbivores. Ces molécules de défense chez la plante sont utilisées en médecine pour soigner un certain nombre d’affections comme des maladies de la peau (bergaptène, utilisé contre le psoriasis), ou le traitement de cancers (xanthotoxine contre le lymphome T). L’équipe Métabolites Secondaires du Laboratoire Agronomie et Environnement cherche à caractériser au niveau moléculaire et enzymatique l’ensemble des réactions impliquées dans la voie de biosynthèse et ainsi à comprendre quels sont les processus évolutifs mis en œuvre dans leur apparition au sein des 4 familles de plantes phylogénétiquement très distinctes. Des éléments de réponses ont été apportés par l’identification de la psoralène synthase (Larbat et al JBC 2007) et de l’angélicine synthase (Larbat et al JBC 2009), enzymes appartenant à la famille des P450.
Le projet vise à la caractérisation de l’ensemble des enzymes de cette voie de biosynthèse particulière par la construction d’une banque BAC de panais et le repérage et séquençage des BACs porteurs des gènes recherchés. Les connaissances générées permettront de révéler le fonctionnement complet de la voie de biosynthèse des phénylpropanoïdes et de comprendre la manière dont les plantes appartenant à des familles très distinctes ont pu évoluer pour s’adapter à un environnement hostile en produisant le même type de molécules. A terme, cela pourrait également permettre par des approches de génie métabolique de moduler/améliorer la synthèse des furocoumarines dans des plantes transgéniques et ainsi générer des plantes transformées dédiées à la production de ces molécules à usage thérapeutique.
Responsable: Sonia VAUTRIN
Implication du CNRGV :
Publication issue du projet:
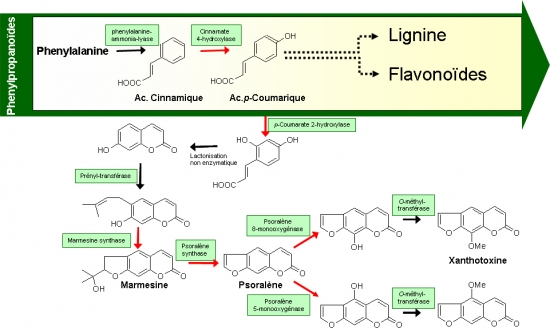
Représentation schématique de la voie de biosynthèse des furocoumarines. Les flèches rouges correspondent à des étapes catalytiques P450 dépendantes

Identification of allelic copies of loci that control traits of economic importance and analysis of its variation between hom(e)ologous chromosomes in sugarcane (Saccharum spp.)
Identification of allelic copies of loci that control traits of economic importance and analysis of its variation between hom(e)ologous chromosomes in sugarcane (Saccharum spp.)
Voir plus
Project coordinator:
Anete Pereira de Souza
Depto de Biologia Vegetal (DBV)
Instituto de Biologia (IB)
Lab. Análise Genética Molecular
Centro de Biol Mol e Eng Gen (CBMEG)
Univ. Est de Campinas (UNICAMP)
Fone: 19-3521-1132
Fax:19-3521-1089
Website: http://www.cbmeg.unicamp.br/
Abstract:
The aim of this project is the caracterization of five genomic regions associated with genes of interest to reveal the allelic organization among different hom(e)ologous regions; and also to identify markers (SSR and SNP) through the sequencing. These markers will be integrated to the map developed by RIDESA, IAC and UNICAMP-ESALQ/USP. To meet this goal two BAC libraries will be constructed for two genotypes of sugarcane in collaboration with CNRGV-INRA. BAC libraries will be organized as 3D pool for one genotype and as macroarrays to find positive clones carrying these five genes. In addition, twenty BAC clones will be sequenced and analyzed.
At UNICAMP, the Center for Molecular Biology and Genetic Engineering, several projects have been developed related to genetic mapping using molecular markers in sugarcane. Therefore these 2 new BAC libraries will also be an essential tool to complement and accelerate the development of genetic maps, facilitating the localization of quantitative traits in them, thus contributing to the improvement of new varieties.
CNRGV involvement:
Responsible: Sonia Vautrin

Breedwheat : Developing new wheat varieties for sustainable agriculture
Developing new wheat varieties for sustainable agriculture.
Voir plus
The project aims at strengthening the competitiveness of the French breeding sector while addressing the societal demand for sustainability, quality and safety. Bringing together 26 partners from the public and private research and breeding sectors, it has a total budget of €34 million for 9 years.
Led by Catherine Feuillet, Research Director at INRA Clermont-Theix, Breedwheat will combine structural and functional genomics, genetics, and ecophysiology with high throughput phenotyping and genotyping to identify markers and genes underlying yield and quality traits under abiotic and biotic stress. Moreover, the project will characterize and tap unexploited genetic resources to expand the diversity of French gene banks. Finally, new breeding methods will be developed and evaluated for their socio-economic impact. .This project will enhance the sustainability of wheat production through the development of new varieties that are more robust and less demanding in water and fertilizer, towards a more environmentally friendly agriculture that is adapted to climate change.
Breedwheat will reinforce the French position in the “Wheat-Global Alliance” for world food security led by the international research centers CIMMYT and ICARDA. It will also enable new collaborations with other large scale national projects in the framework of the new international “Wheat Initiative” for the coordination of wheat research, supported by the G20, co-led by INRA and launched on September 15th, 2011 at the French Ministry of Agriculture.
Breedwheat is the 1st French Stimulus Initiative project coordinated by INRA in the category “Biotechnologies and Bioresources” to be launched. Announced on February 23rd, 2011 by the French Minister of Higher Education and Research and the French Minister of Agriculture, Breedwheat is one of the five laureates of the “Biotechnologies and Bioresources” call for proposals. The project represents a total investment of €34 million for 9 years, of which €9 million were granted by the French Stimulus Initiative through the French National Research Agency (ANR). The project gathers the competencies of 26 French partners, including 11 private companies, to develop and use efficient genome sequence-based tools and new methodologies for breeding wheat varieties with improved quality, sustainability, and productivity.
Project coordinator:
INRA - UMR 1095 Génétique, Diversité, Ecophysiologie des Céréales (GDEC)
5, chemin de Beaulieu
63100 Clermont-Ferrand
France
![]()
Project partners:
The project involves 26 French partners, including 11 private companies.
CNRGV's responsible
CNRGV involvement:

Towards the understanding of the mechanisms involved in frost tolerance in pea.
Towards the understanding of the mechanisms involved in frost tolerance in pea.
Voir plus
In collaboration with :
Bruno Delbreil
UMR USTL INRA 1281, Stress abiotiques et différenciation des végétaux cultivés
Université des Sciences et technologies de Lille, Bâtiment SN2
59650 Villeneuve d’Ascq
France
Phone: +33 320434022
E-mail: bruno.delbreil@univ-lille1.fr
Isabelle Lejeune
UMR USTL INRA 1281, Stress abiotiques et différenciation des végétaux cultivés
80200 Estrées-Mons
France
Phone: +33 322857507
E-mail: Lejeune@mons.inra.fr
Nasser Bahrman
UMR USTL INRA 1281, Stress abiotiques et différenciation des végétaux cultivés
Université des Sciences et technologies de Lille, Bâtiment SN2
59650 Villeneuve d’Ascq
France
Phone: +33 322857854
E-mail: nasser.bahrman@mons.inra.fr
Abstract :
Dry pea is an important field crop in France. The sowing of dry pea in autumn allows a longer life cycle than in a spring sowing, associated with a higher biomass production that would help to improve and stabilize this crop productivity. However, to release winter varieties, winter survival has still to be improved and particularly the freezing tolerance component. On pea, the genetic determinism of frost tolerance has been studied. Three major QTLs have been mapped on linkage groups 3, 5 and 6.
Pea BAC-library are important genomic tools suitable for physical mapping and map-based cloning. This library is being screened with probes corresponding to markers in the QTL interval. The physical map of the corresponding region is under construction and BACs covering the reduced confidence interval of the QTL will be sequenced. This approach that combine fine mapping and positional cloning will lead us to identify potential candidate genes underlying the three freezing tolerance pea QTLs.
Recent publications :
E. Dumont, N. Bahrman, E. Goulas, B. Valot, H. Sellier, JL. Hilbert, C. Vuylsteker, I. Lejeune-Hénaut, B. Delbreil. A proteomic approach to decipher chilling response from cold acclimation in pea (Pisum sativum L.) Plant Sci. 2011 (180-1) pp 86-98
E. Dumont, V. Fontaine, C. Vuylsteker, H. Sellier, S. Bodèle, N.Voedts, R.Devaux, M.Frise, K. Avia, JL Hilbert, N.Bahrman, E.Hanocq, I.Lejeune-Hénaut, B.Delbreil. Association of sugar content QTL and PQL with physiological traits relevant to frost damage resistance in pea under field and controlled conditions. TAG 2008 (118-8) pp 1561-1571

CNRGV's responsible: Sonia Vautrin

Screening the R570 sugarcane BAC library to identify BAC clones that underlie QTL of agronomic importance
In the last five years a consortium of scientists from a number of countries including Australia have grouped together to form the Sugarcane Genome Sequencing Initiative (SUGESI). The goal of this consortium is to generate a combined monoploid genom e sequence of sugarcane.
Voir plus
Project
Dr Karen Aitken
CSIRO Plant Industry
Queensland Bioscience Precinct
306 Carmody Road
St Lucia
4068
QLD Australia
Abstract
Over the last decade a number of genetic maps have been constructed and quantitative trait loci (QTL) studies carried out for agronomically important traits. These have included traits related to sugar content (Brix, Pol), as well as yield and biomass related traits such as stem diameter, height and stalk number. In parallel a large association study using the parental population of the Australian sugarcane breeding program has also been conducted targeting disease resistance. These studies have identified many markers that contribute to the phenotypic variation of these traits. The next step is to determine the genetic structure of these regions, and which genes or regulatory elements underlie these QTL. In the last five years a consortium of scientists from a number of countries including Australia have grouped together to form the Sugarcane Genome Sequencing Initiative (SUGESI). The goal of this consortium is to generate a combined monoploid genome sequence of sugarcane. The Australian contribution to this consortium is sequenced BAC clones from the R570 BAC library that underlie the QTLs identified in the numerous studies. The R570 BAC library will be screened at INRA-CNRGV to identify the BAC clones that underlie these QTL of agronomic importance.
The aims of this project are:
1) To establish a strategy to identify which QTL regions to target.
QTL regions were selected that had been identified at a significance level of less than p=0.001 and contained more than one marker. Ideally, the selected QTL should also have been identified in more than one population. In the next stage, sets of linked markers were selected to test across the whole QTL region. The selected markers were in most cases SNP or DArT markers with a known sequence. The sequenced markers were BLASTn aligned to the sorghum genome to determine the abundance of the sequence. Only those markers that aligned to a single location in sorghum were selected to screen the BAC clones and preference was given to those that also showed alignment to the sugarcane EST collection.
2) Screen the R570 BAC library with the selected markers linked to QTL.
Two different approaches: macroarray membrane hybridisation and qPCR using 3-D BAC pools will be used to screen the library. In total 112 primer pairs were developed to screen the R570 BAC library. These will be used to identify BAC clones which will then be characterised and ultimately sequenced to identify genes in these regions. Once the sequence is obtained a major outcome will be the development of robust markers, closely linked to causal genes, for use in the Australian sugarcane breeding program.
CNRGV's responsible
Sonia Vautrin

Genomics of Sunflower
The CNRGV is the partner of the genome sequencing project involving Canada, USA and France. In this project, the CNRGV will be in charge of the constructions of the new sunflower BAC libraries from the genotype HA412-HO. 3 BAC libraries will be constructed using 3 different enzymes (HindIII, EcoRI, and BamHI) in order to reach a 12 to 14 x coverage of the sunflower genome.
Voir plus
The Compositae is one of the largest and most economically important families of flowering plants and includes a diverse array of food crops, horticultural crops, medicinals, and noxious weeds (Dempewolf et al. 2008). Despite its size and economic importance, genomic characterization has lagged behind that of comparable groups such as the grass, legume, crucifer, and tomato families. In particular, the lack of a reference sequence for the Compositae impedes research and improvement efforts. We will dramatically enhance Compositae genomic resources by sequencing the genome of cultivated sunflower ( Helianthus annuus), the most important crop in the family. In addition to its global importance as an oil crop, sunflower has tremendous potential for cellulosic biomass production, both as a primary source and via the residue of oilseed production. Thus, the proposed large-scale sequencing effort will be accompanied by the development of genomic resources, genetic stocks, and knowledge for manipulating agronomically important traits in sunflower hybrid breeding programs.
The scientific objectives of the project are to :
1) Extend the genetic map for sunflower;
2) Construct a physical map of the sunflower genome based on fingerprinting and end-sequencing of BAC clones from deep-coverage libraries;
3) Generate a reference sequence for sunflower using a hybrid strategy involving whole genome shotgun sequencing (40x depth) and sequencing of BAC pools (20x depth) with nextgeneration sequencing platforms;
4) Determine the genetic basis of agriculturally important traits using an association mapping approach; and
5) Develop a xylem EST database and determine the genetic basis of cellulosic biomass traits.
The project will create an unparalleled resource for sunflower and the Compositae and yield a wealth of data for functional and comparative genomic analyses and agricultural, biological, and environmental research. Candidate gene and SNP discovery will facilitate public and private crop improvement efforts, as well as the ongoing development of sunflower as a biofuel. With climate change, drought tolerant Compositae species are likely to become increasingly important as food and biofuel crops in Canada and elsewhere, as well as biological factories for the production of pharmaceuticals (Maloney 2000). Also because many of the most noxious weeds in Canada are Compositae species (e.g., thistles, napweeds, and ragweeds), a reference genome for the family will aid ongoing efforts to understand and control these invasive species. Finally, our results will allow us to test a new theory about the role of genomic redundancy in the establishment of chromosomal rearrangements and possibly solve a longstanding mystery in plant genome evolution.
Website :
http://www.sunflowergenome.org/
Partners :
Loren Rieseberg
Botany Department University of British Columbia
3529-6270 University Blvd Vancouver,
B.C. V6T 1Z4
CANADA
Phone: 604-827-4540
Fax: 604-822-6089
LIPM "SUNFLOWER" Team
INRA
Chemin de Borde Rouge
31326 Castanet Tolosan
FRANCE
Tel : 05 61 28 54 58
Mob : 06 73 69 23 72
Steven J. Knapp
Professor and Georgia Research Alliance Eminent Scholar
Institute of Plant Breeding, Genetics, and Genomics
Center for Applied Genetic Technologies
111 Riverbend Road
The University of Georgia
Athens, Georgia 30602
USA
Tel: (706) 542-4021
Fax (706) 583-8120
E-mail: sjknapp@uga.edu
Publications related to the project :
CNRGV's responsible

The Sugarcane Genome Sequencing Initiative (SUGESI): Strategies for Sequencing a Highly Complex Genome
Sugarcane cultivars derive from recent interspecific hybrids obtained by crossing Saccharum officinarum and Saccharum spontaneum and represent relevant feedstock used worldwide for biofuel production.
Voir plus
Sugarcane cultivars derive from recent interspecific hybrids obtained by crossing Saccharum officinarum and Saccharum spontaneum and represent relevant feedstock used worldwide for biofuel production. The challenge in a sugarcane sequencing project is the size (10 Gb) and complexity of its genome structure that is highly polyploid and aneuploid with a complete set of homo(eo)logous genes predicted to range from 8 to 10 copies (alleles). Although sugarcane’s monoploid genome is about 1 Gb, its highly polymorphic nature represent another significant challenge for obtaining a genuine assembled monoploid genome. A sugarcane R570 BAC library whose construction was funded by ICSB is available for sequencing. The genome coverage of this library is believed to be around 1,3x suggesting that recovery of all alleles would not be always efficient. In any case, BAC screening can be undertaken using a fast and efficient 3D-pool approach which has been developed at CNRGV followed by PCR amplification of genes of interest.
The Sugarcane Genome Sequencing Initiative – A particularly attractive initial strategy that lies at the intersection of the common interests of virtually all of the sugarcane research community is to capture much of the gene-rich recombinationally-active euchromatin. The Sugarcane Genome Sequencing Initiative (SUGESI) was envisaged to join efforts to produce a reference sequence of a sugarcane cultivar ( Saccharum hybrid) and ancestor genotypes ( Saccharum officinarum and Saccharum spontaneum) using a combination of approaches but mainly focused on BAC sequencing. The SUGESI Consortium aggregates researchers from Australia, Brazil, France, South Africa and United States. The SUGESI Initiative intends to make sequences public as soon as minimum assemblies permit.
Publications related to the project :
List of partners:
Dr Marie-Anne Van Sluys, Professor of Botany GaTE Lab (Genomics and Transposable elements)
Departamento de Botânica-IBUSP
rua do Matao, 277 05508-900;
São Paulo, SP, BRASIL
Dr Glaucia Souza
Associate Professor
BIOEN Program Coordinator Instituto de Química
Universidade de São Paulo
Dr Angélique D'Hont
CIRAD, UMR 1098 DAP Equipe "Structure et évolution des génomes" TAA96/03,
Avenue Agropolis
34398 Montpellier cedex 5, FRANCE
Dr Andrew Paterson
Plant Genome Mapping Laboratory Center for Applied Genetic Technologies
111 Riverbend Road, Athens, GA 30606
Dr Ray Ming
University of Illinois at Urbana-Champaign
1201 W. Gregory Drive 148 ERML, MC-051
Dr Robert Henry
Southern Cross University
Centre for Plant Conservation Genetics Bioenergy Research Institute/Lismore, NSW Australia
Dr Rosanne Casu
Queensland Bioscience Precinct -
St Lucia 306 Carmody Road
St Lucia QLD 4067 Australia
Dr Bernard Potier
South African Sugarcane Research Institute
Private Bag X02 Mount Edgecombe
4300 South Africa
CNRGV's responsible
Screening of wheat genomic BAC libraries with specific primers for the TdDRF1 (Triticum durum Dehydration responsive Factor 1) gene
Drought is one of the most severe abiotic stresses limiting crop productivity and our understanding, at the molecular level, of crop response to water stress is further increasing.
Voir plus
In collaboration with:
Website : http://www.enea.it/com/ingl/default.htm
Laboratory Leader: Dr. P. Galeffi
Principal Researcher: Dr. A. Latini
Abstract
The Triticum durum Dehydration Responsive Factor 1 gene, TdDRF1, belonging to the DREB gene family, is expressed in response to dehydration and encodes for abiotic stress responsive transcription factors that impart stress endurance to the plants (Latini et al. 2007). Recently, the expression profile of this gene was finely examined in several wheat varieties in both greenhouse and in field conditions, during a water stress time-course (Latini et al. 2008). All obtained data spotlighted that the targeting of this gene could help the selection of cultivars for improving plant tolerance to drought.
The fine mapping of this gene, determining the exact locus position on chromosomes will be very interesting and useful. Actually, it is already established that homeologous loci of this gene exist in 1A, 1B chromosomes of durum wheat and 1A, 1B and 1D chromosomes of bread wheat; in addiction, there are some evidences about the existence of some paralogous gene copies and also pseudogenes without introns.
The collaboration with the CNRGV is highly fruitful, because it holds the most relevant BAC genomic DNA collections of wheat. In addiction to several collections of bread wheat, at CNRGV, durum wheat (Cenci et al. 2003) and chromosome-specific (such as 1D, 4D, 6D and 1AS or 1AL of hexaploid wheat – Janda et al. 2004) genomic BAC libraries are also available.
Actually, the bread wheat Chinese Spring BAC library (Tae-B-CS) was screened by both PCR on pools and microarray hybridization techniques with primers specific for the TdDRF1 sequence. A durum wheat library is still under screening. Several clones containing the target sequence were identified and are being characterized by restriction analyses, chromosome walking and complete BAC sequencing.
From the positive BAC clones, the upstream and downstream TdDRF1 gene regions will be sequenced, allowing the identification of the promoter and other regulatory sequences (enhancers, alternative splicing signals, and so on). Furthermore, the complete sequence of a selected BAC clone will reveal if some other dehydration-responsive gene is located next to the target locus and this information will be precious for developing applications in molecular assisted breeding programs aimed to the release of drought tolerant wheat cultivars.
- Latini A, Rasi C, Sperandei M, Cantale C, Iannetta M, Dettori M, Ammar K, Galeffi P: Identification of a DREB-related gene in Triticum durum and its expression under water stress conditions. Annals of Applied Biology (2007), 150: 187-195.
- Latini A, Sperandei M, Sharma S, Cantale C, Iannetta M, Dettori M, Ammar K, Galeffi P: Molecular analyses of a dehydration-related gene from the DREB family in durum wheat and triticale. In Biosaline Agriculture and High Salinity Tolerance. Published by Birkhäuser and Verlag. Edited by Abdelly C, Öztürk M, Ashraf M, Grignon C. (2008), 286-295.
- Cenci a, Chantret N, Kong X, Gu Y, Anderson OD, Fahima T, Distelfeld, Dubcovsky J: Construction and characterization of a half million clone BAC library of durum wheat ( Triticum turgidum ssp. durum). Theoretical and Applied Genetics (2003), 107: 931-939.
- Janda J, Bartoš J, Šafář J, Kubaláková M, Valárik M, Čihalíková J, Šimková H, Caboche M, Sourdille P, Bernard M, Chalhoumb B, Doležel J: Construction of a subgenomic BAC library specific for chromosomes 1D, 4D and 6D of Hexaploid wheat. Theoretical and Applied Genetics (2004) 109: 1337-1345.
This collaboration was also supported by a COST-STSM-FA0604 grant, given to A. Latini.
CNRGV involvement
Responsible : Elisa PRAT/ Sonia VAUTRIN
- Macroarray production/ screening
- 2D DNA pools screening
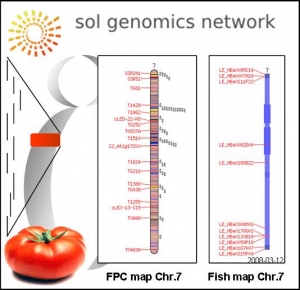
Séquençage du génome de la tomate
Gestion des banques BAC de la tomate et définition du minimum tilling path pour le séquençage du chromosome 7
Voir plus
Gestion des banques BAC de la tomate et définition du minimum tilling path pour le séquençage du chromosome 7
![]()
Projet développé dans le cadre du Solanaceae Genome Network
En collaboration avec
Site internet: http://gbf.ensat.fr/gbf_projects.html
Résumé
Un projet international, International Solanaceae Genome Initiative, s’est mis en place dans le but de séquencer le génome de la tomate. La stratégie qui a été adoptée consiste à séquencer les régions riches en gènes. Elle découle de résultats qui montrent que chez la tomate la grande majorité des séquences géniques se trouvent concentrée dans des régions euchromatiques contiguës et constituant environ 25% des 950 Mb du génome de la tomate. La démarche choisie consiste à identifier et à séquencer un minimum tilling path de clones BAC présent dans ces 220Mb d’euchromatine. Le génome de la tomate est constitué de 12 chromosomes. Chaque pays partenaire participe à cet effort en prenant en charge le séquençage d’un ou plusieurs chromosomes ( http://www.sgn.cornell.edu/about/tomato_sequencing.pl ). En France, le laboratoire GBF de Monder Bouzayen, est responsable du séquençage du chromosome 7.
Résultats
La première version de la séquence complète du génome de la tomate vient d’être obtenue par le consortium international et une première version de ces données est disponible sur le site SGN ( http://solgenomics.net/tomato/ ).
Techniques utilisées par le CNRGV
Responsable : Sonia VAUTRIN
- Criblage sur Macroarrays
- Production de pools en 3D DNA: banque SL_MboI et SL_HindIII
- Criblage sur pools
TriticaeGenome : Vers la caractérisation du génome du blé et de l'orge
Vers la caractérisation du génome du blé et de l'orge
Voir plus
![]()
Seventh framework programmeFood Agriculture and Fisheries, Biotechnology
Coordinateur du projet
Résumé
Depuis de nombreuses années, la grandeur et la complexité des génomes du blé, de l'orge et du seigle ont ralenti le développement des approches génomiques et de leurs applications dans l’amélioration de la production et la qualité des récoltes. Cependant, récemment, de nouvelles approches technologiques plus efficaces ont été développées et de nouvelles ressources on été générées qui permettent que des programmes de génomiques robustes puissent être envisagés pour les Triticeae.
TriticeaeGenome est conçu pour accomplir des progrès significatifs dans la génomique des Triticeae et soutenir la reproduction efficace de variétés améliorées pour l'agriculture européenne. Ce projet sera développé par :
Triticeaegenome est mis en place comme une contribution majeure aux efforts de consortiums internationaux dans le fait de construire des cartes physiques d'orge et de blé hexaploïde pour l’amélioration de l’agriculture, l'accélération de l'isolement de gènes et QTL et la mise en place de bases solides pour les prochains programmes de séquençage de ces génomes.
Triticeaegenome donnera des informations et des outils originaux aux éleveurs et aux scientifiques pour une meilleure compréhension de l'organisation des génomes des Triticeae, leur évolution et leur fonction, en fournissant une meilleure compréhension de la biologie de ces plantes cultivées essentielles.
liens utiles :
http://tritigen.ari.gov.cy/
http://www.wheatgenome.org/
http://urgi.versailles.inra.fr/Projects/TriticeaeGenome
Implications du CNRGV
Responsable : Arnaud Bellec/ Sonia Vautrin
- Conservation de banques BAC chromosome spécifique du blé.
- Rearrangement du Minimum Tilling Path
- Production de pools 3D sur les banques BACs chromosomes spécifiques Blé et sur la banque BAC Orge
Laboratoires partenaires
PROMOSOL
|
| Etude de la résistance du tournesol au mildiou ( Plasmopara halstedii) |

Voir plus
![]()
Etude de la Résistance du tournesol au mildiou ( Plasmopara halstedii )
Coordinateur du projet
Résumé
Le mildiou du Tournesol causé par Plasmopara Halstedii est l’une des affections les plus potentiellement dangereuses en Europe. Son développement est favorisé par les pluies de printemps, dont la fréquence peut s’accroître du fait de la tendance à encourager les semis précoces pour faire bénéficier la culture, à des fins d’augmentation de la productivité, d’un apport en eau plus important. De façon concomitante au développement de culture, les variétés ont apporté des facteurs de résistance très efficaces à court-moyen terme, mais ceux-ci ont été peu à peu contournés par l’apparition de nouvelles races du parasite. Ce projet se propose de développer des outils et des connaissances dont la prise en compte globale permettra d’élaborer des stratégies de mise en œuvre d’une résistance durable, par la création et l’exploitation de ressources génétiques de Plasmopara halstedii en vue d’analyser la variabilité naturelle des populations du parasite et de progresser dans la connaissance et l’identification des pathotypes, l’identification et la cartographie de nouvelles sources de résistance race spécifique déjà recensées au sein d’un vaste ensemble de ressources génétique du genre Helianthus, la cartographie fine et les premières étapes du clonage positionnel d’un QTL de résistance quantitative partielle, Et la construction d’une ressource génétique Tournesol qui permettra ultérieurement l’analyse plus fine de l’interaction hôte-parasite dans l’espace et dans le temps.
Liens:
http://lipm-helianthus.toulouse.inra.fr/dokuwiki/doku.php?id=start

Implications du CNRGV
Responsable : Sonia VAUTRIN
Laboratoires partenaires
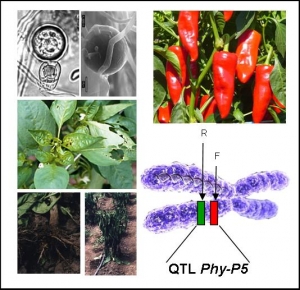
PHYTOSOL-2
Validation fonctionnelle de gènes candidats pour un QTL à spectre large agissant sur la résistance aux Phytophthora chez les Solanacées
Voir plus
Validation fonctionnelle de gènes candidats pour un QTL à spectre large agissant sur la résistance aux Phytophthora chez les Solanacées
![]()
![]()
Projet ANR - Réseau de Génomique Végétale - Génoplante 2010
Coordinateur du projet :
Dr. Véronique Lefebvre
INRA-UR1052-GAFL
Caractérisation Fonctionnelle des interactions Plantes-Bioagresseurs
BP94 - 84140 Montfavet - France
Email: veronique.lefebvre@avignon.inra.fr
Résumé :
Les Phytophthora sont les parasites les plus dévastateurs des Dicotylédones. La lutte chimique a été interdite par la commission européenne. Les résistances monogéniques sont fréquemment contournées. Bien que les résistances polygéniques aient été démontrées plus durables, leurs bases moléculaires demeurent inconnues.
Nous avons pour objectif de déterminer la nature moléculaire d’un QTL impliqué dans la résistance à plusieurs espèces de Phytophthora chez les Solanacées. Ce QTL, Phy-P5, confère un niveau élevé de résistance à plusieurs isolats de P. capsici chez le piment et montre une position colinéaire avec des QTL de résistance à P. infestans chez la tomate et la pomme de terre. L’analyse génétique du locus Phy-P5 a permis d’identifier deux QTL finement liés dans 0.57 cM. Deux clones BAC contenant chacun un des deux gènes sous-jacents à Phy-P5 seront séquencés, annotés, et ancrés à la carte génétique en 2007, afin de sélectionner les gènes candidats.
Les objectifs du projet PHYTOSOL-2 sont de valider les gènes candidats identifiés pour le QTL Phy-P5, et de développer un système modèle facilitant la caractérisation fonctionnelle des gènes impliqués dans l’interaction Phytophthora/Solanacées.
La variation naturelle dans les séquences des gènes candidats sera analysée chez les lignées parentales et plusieurs accessions de piment (EcoTILLING). Nous identifierons les domaines fonctionnels des gènes par analyse de recombinants intragéniques et séquençage d’allèles chimères. La validation des gènes candidats sera réalisée par transformation via Agrobacterium rhizogenes chez le piment, et par transformation stable chez la tomate. L’expression spatio-temporelle des gènes identifiés et les cascades de transduction du signal induites seront déterminées chez le piment et/ou la tomate. La validation fonctionnelle des gènes impliqués dans les réponses induites sera réalisée par génétique inverse en exploitant la plateforme TILLING de la tomate. Enfin, les résultats obtenus seront exploités pour améliorer la sélection de plantes résistantes aux Phytophthora.
Techniques utilisées par le CNRGV :
Responsable : Sonia Vautrin
- Construction de pools piment "Capsicum annum HD208" en deux dimensions
- Criblage des pools d' ADN, par PCR Quantitative en temps réel
- Séquençage des BACs Ends
- Agencement des clones BACs sur la carte physique du QTL phy-P5
Laboratoires partenaires :







Repeat-length variation in a wheat cellulose synthase-like gene is associated with altered tiller number and stem cell wall composition.
J Exp Bot. doi: 10.1093/jxb/erx051
Voir plus
Authors :
Hyles J, Vautrin S, Pettolino F, MacMillan C, Stachurski Z, Breen J, Berges H, Wicker T, Spielmeyer W.
J Exp Bot. doi: 10.1093/jxb/erx051
Abstract :
The tiller inhibition gene (tin) that reduces tillering in wheat (Triticum aestivum) is also associated with large spikes, increased grain weight, and thick leaves and stems. In this study, comparison of near-isogenic lines (NILs) revealed changes in stem morphology, cell wall composition, and stem strength. Microscopic analysis of stem cross-sections and chemical analysis of stem tissue indicated that cell walls in tin lines were thicker and more lignified than in free-tillering NILs. Increased lignification was associated with stronger stems in tin plants. A candidate gene for tin was identified through map-based cloning and was predicted to encode a cellulose synthase-like (Csl) protein with homology to members of the CslA clade. Dinucleotide repeat-length polymorphism in the 5'UTR region of the Csl gene was associated with tiller number in diverse wheat germplasm and linked to expression differences of Csl transcripts between NILs. We propose that regulation of Csl transcript and/or protein levels affects carbon partitioning throughout the plant, which plays a key role in the tin phenotype.
Long Read Sequencing Technology to Solve Complex Genomic Regions Assembly in Plants.
Journal of Next Generation Sequencing & Applications.
Voir plus
Authors :
Arnaud Bellec, Audrey Courtial, Stephane Cauet, Nathalie Rodde, Sonia Vautrin, Genseric Beydon, Nadege Arnal, Nadine Gautier, Joelle Fourment, Elisa Prat, William Marande, Yves Barriere and Helene Berges.
Journal of Next Generation Sequencing & Applications.
Abstract :
Background:
Numerous completed or on-going whole genome sequencing projects have highlighted the fact that obtaining a high quality genome sequence is necessary to address comparative genomics questions such as structural variations among genotypes and gain or loss of specific function. Despite the spectacular progress that has been made in sequencing technologies, obtaining accurate and reliable data is still a challenge, both at the whole genome scale and when targeting specific genomic regions. These problems are even more noticeable for complex plant genomes. Most plant genomes are known to be particularly challenging due to their size, high density of repetitive elements and various levels of ploidy. To overcome these problems, we have developed a strategy to reduce genome complexity by using the large insert BAC libraries combined with next generation sequencing technologies.
Results:
We compared two different technologies (Roche-454 and Pacific Biosciences PacBio RS II) to sequence pools of BAC clones in order to obtain the best quality sequence. We targeted nine BAC clones from different species (maize, wheat, strawberry, barley, sugarcane and sunflower) known to be complex in terms of sequence assembly. We sequenced the pools of the nine BAC clones with both technologies. We compared assembly results and highlighted differences due to the sequencing technologies used.
Conclusions:
We demonstrated that the long reads obtained with the PacBio RS II technology serve to obtain a better and more reliable assembly, notably by preventing errors due to duplicated or repetitive sequences in the same region.
Link :
A Metabolic Gene Cluster in the Wheat W1 and the Barley Cer-cqu Loci Determines β-Diketone Biosynthesis and Glaucousness.
Plant Cell. 2016 May 25. pii: tpc.00197.2016.
Voir plus
Authors :
Hen-Avivi S, Savin O, Racovita R, Lee WS, Adamki N, Malitsky S, Almekias-Siegl E, Levy M, Vautrin S, Bergès H, Friedlander G, Kartvelishvily E, Ben-Zvi G, Alkan N, Uauy C, Kanyuka K, Jetter R, Distelfeld A, Aharoni A.
Plant Cell. 2016 May 25. pii: tpc.00197.2016.
Abstract :
The glaucous appearance of wheat and barley plants, that is the light bluish-grey look of flag leaf, stem, and spike surfaces, results from deposition of cuticular β-diketone wax on their surfaces; this phenotype is associated with high yield, especially under drought conditions. Despite extensive genetic and biochemical characterization, the molecular genetic basis underlying the biosynthesis of β-diketones remains unclear. Here we discovered that the wheat W1 locus contains a metabolic gene cluster mediating β-diketone biosynthesis. The cluster comprises genes encoding proteins of several families including type-III polyketide synthases, hydrolases, and cytochrome P450s related to known fatty acid hydroxylases. The cluster region was identified in both genetic and physical maps of glaucous and glossy tetraploid wheat, demonstrating entirely different haplotypes in these accessions. Complementary evidence obtained through gene silencing in planta and heterologous expression in bacteria supports a model for a β-diketone biosynthesis pathway involving members of these three protein families. Mutations in homologous genes were identified in the barley eceriferum mutants defective in β-diketone biosynthesis, demonstrating a gene cluster also in the β-diketone biosynthesis Cer-cqu locus in barley. Hence, our findings open new opportunities to breed major cereal crops for surface features that impact yield and stress response.
Suppressed recombination and unique candidate genes in the divergent haplotype encoding Fhb1, a major Fusarium head blight resistance locus in wheat.
Theor Appl Genet. 2016 DOI: 10.1007/s00122-016-2727-x.
Voir plus
Authors :
Schweiger W, Steiner B, Vautrin S, Nussbaumer T, Siegwart G, Zamini M, Jungreithmeier F, Gratl V, Lemmens M, Mayer KF, Bergès H, Adam G, Buerstmayr H.
Theor Appl Genet. 2016 DOI: 10.1007/s00122-016-2727-x.
Abstract :
Fine mapping and sequencing revealed 28 genes in the non-recombining haplotype containing Fhb1 . Of these, only a GDSL lipase gene shows a pathogen-dependent expression pattern. Fhb1 is a prominent Fusarium head blight resistance locus of wheat, which has been successfully introgressed in adapted breeding material, where it confers a significant increase in overall resistance to the causal pathogen Fusarium graminearum and the fungal virulence factor and mycotoxin deoxynivalenol. The Fhb1 region has been resolved for the susceptible wheat reference genotype Chinese Spring, yet the causal gene itself has not been identified in resistant cultivars. Here, we report the establishment of a 1 Mb contig embracing Fhb1 in the donor line CM-82036. Sequencing revealed that the region of Fhb1 deviates from the Chinese Spring reference in DNA size and gene content, which explains the repressed recombination at the locus in the performed fine mapping. Differences in genes expression between near-isogenic lines segregating for Fhb1 challenged with F. graminearum or treated with mock were investigated in a time-course experiment by RNA sequencing. Several candidate genes were identified, including a pathogen-responsive GDSL lipase absent in susceptible lines. The sequence of the Fhb1 region, the resulting list of candidate genes, and near-diagnostic KASP markers for Fhb1 constitute a valuable resource for breeding and further studies aiming to identify the gene(s) responsible for F. graminearum and deoxynivalenol resistance.
The wheat Sr50 gene reveals rich diversity at a cereal disease resistance locus
Nature Plants. Article number:15186 (2015). DOI:10.1038/nplants.2015.186
Voir plus
Nature Plants. Article number:15186 (2015). DOI:10.1038/nplants.2015.186
Authors :
Rohit Mago, Peng Zhang, Sonia Vautrin, Hana Šimková, Urmil Bansal, Ming-Cheng Luo, Matthew Rouse, Haydar Karaoglu, Sambasivam Periyannan, James Kolmer, Yue Jin, Michael A. Ayliffe, Harbans Bariana, Robert F. Park, Robert McIntosh, Jaroslav Doležel, Hélène Bergès, Wolfgang Spielmeyer, Evans S. Lagudah, Jeff G. Ellis, Peter N. Dodds
Abstract :
We identify the wheat stem rust resistance gene Sr50 (using physical mapping, mutation and complementation) as homologous to barley Mla, encoding a coiled-coil nucleotide-binding leucine-rich repeat (CC-NB-LRR) protein. We show that Sr50 confers a unique resistance specificity different from Sr31 and other genes on rye chromosome 1RS, and is effective against the broadly virulent Ug99 race lineage. Extensive haplotype diversity at the rye Sr50 locus holds promise for mining effective resistance genes.
Link :
The first insight into the Salvia (lamiaceae) genome via BAC library construction and high-throughput sequencing of target BAC clones.
(2015). Pak. J. Bot., 47(4): 1347-1357.
Voir plus
Authors :
Da Cheng Hao, Sonia Vautrin, Chi Song, Ying Jie Zhu, Helene Berges, Chao Sun and Shi Lin Chen
Pak. J. Bot., 47(4): 1347-1357, 2015.
Abstract :
Salvia is a representative genus of Lamiaceae, a eudicot family with significant species diversity and population adaptibility. One of the key goals of Salvia genomics research is to identify genes of adaptive significance. This information may help to improve the conservation of adaptive genetic variation and the management of medicinal plants to increase their health and productivity. Large-insert genomic libraries are a fundamental tool for achieving this purpose. We report herein the construction, characterization and screening of a gridded BAC library for Salvia officinalis (sage). The S. officinalis BAC library consists of 17,764 clones and the average insert size is 107 Kb, corresponding to similar to 3 haploid genome equivalents. Seventeen positive clones (average insert size 115 Kb) containing five terpene synthase (TPS) genes were screened out by PCR and 12 of them were subject to Illumina HiSeq 2000 sequencing, which yielded 28,097,480 90-bp raw reads (2.53 Gb). Scaffolds containing sabinene synthase (Sab), a Sab homolog, TPS3 (kaurene synthase-like 2), copalyl diphosphate synthase 2 and one cytochrome P450 gene were retrieved via de novo assembly and annotation, which also have flanking noncoding sequences, including predicted promoters and repeat sequences. Among 2,638 repeat sequences, there are 330 amplifiable microsatellites. This BAC library provides a new resource for Lamiaceae genomic studies, including microsatellite marker development, physical mapping, comparative genomics and genome sequencing. Characterization of positive clones provided insights into the structure of the Salvia genome. These sequences will be used in the assembly of a future genome sequence for S. officinalis.
The physical map of wheat chromosome 5DS revealed gene duplications and small rearrangements.
BMC Genomics. 2015 Jun 13;16:453. doi: 10.1186/s12864-015-1641-y.
Voir plus
Authors :
Akpinar BA, Magni F, Yuce M, Lucas SJ, Šimková H, Šafář J, Vautrin S, Bergès H, Cattonaro F, Doležel J, Budak H.
BMC Genomics. 2015 Jun 13;16:453. doi: 10.1186/s12864-015-1641-y.
Abstract :
BACKGROUND:
The substantially large bread wheat genome, organized into highly similar three sub-genomes, renders genomic research challenging. The construction of BAC-based physical maps of individual chromosomes reduces the complexity of this allohexaploid genome, enables elucidation of gene space and evolutionary relationships, provides tools for map-based cloning, and serves as a framework for reference sequencing efforts. In this study, we constructed the first comprehensive physical map of wheat chromosome arm 5DS, thereby exploring its gene space organization and evolution.
RESULTS:
The physical map of 5DS was comprised of 164 contigs, of which 45 were organized into 21 supercontigs, covering 176 Mb with an N50 value of 2,173 kb. Fifty-eight of the contigs were larger than 1 Mb, with the largest contig spanning 6,649 kb. A total of 1,864 molecular markers were assigned to the map at a density of 10.5 markers/Mb, anchoring 100 of the 120 contigs (>5 clones) that constitute ~95 % of the cumulative length of the map. Ordering of 80 contigs along the deletion bins of chromosome arm 5DS revealed small-scale breaks in syntenic blocks. Analysis of the gene space of 5DS suggested an increasing gradient of genes organized in islands towards the telomere, with the highest gene density of 5.17 genes/Mb in the 0.67-0.78 deletion bin, 1.4 to 1.6 times that of all other bins.
CONCLUSIONS:
Here, we provide a chromosome-specific view into the organization and evolution of the D genome of bread wheat, in comparison to one of its ancestors, revealing recent genome rearrangements. The high-quality physical map constructed in this study paves the way for the assembly of a reference sequence, from which breeding efforts will greatly benefit.
Major haplotype divergence including multiple germin-like protein genes, at the wheat Sr2 adult plant stem rust resistance locus.
BMC Plant Biol. 2014. 14(1):1585.
Voir plus
Authors
Mago R, Tabe L, Vautrin S, Imková H, Kubaláková M, Upadhyaya N, Berges H, Kong X, Breen J, Dole El J, Appels R, Ellis J, Spielmeyer W
BMC Plant Biol. 2014. 14(1):1585.
Abstract
The adult plant stem rust resistance gene Sr2 was introgressed into hexaploid wheat cultivar (cv) Marquis from tetraploid emmer wheat cv Yaroslav, to generate stem rust resistant cv Hope in the 1920s. Subsequently, Sr2 has been widely deployed and has provided durable partial resistance to all known races of Puccinia graminis f. sp. tritici. This report describes the physical map of the Sr2-carrying region on the short arm of chromosome 3B of cv Hope and compares the Hope haplotype with non-Sr2 wheat cv Chinese Spring.Results Sr2 was located to a region of 867 kb on chromosome 3B in Hope, which corresponded to a region of 567 kb in Chinese Spring. The Hope Sr2 region carried 34 putative genes but only 17 were annotated in the comparable region of Chinese Spring. The two haplotypes differed by extensive DNA sequence polymorphisms between flanking markers as well as by a major insertion/deletion event including ten Germin-Like Protein (GLP) genes in Hope that were absent in Chinese Spring. Haplotype analysis of a limited number of wheat genotypes of interest showed that all wheat genotypes carrying Sr2 possessed the GLP cluster; while, of those lacking Sr2, some, including Marquis, possessed the cluster, while some lacked it. Thus, this region represents a common presence-absence polymorphism in wheat, with presence of the cluster not correlated with presence of Sr2. Comparison of Hope and Marquis GLP genes on 3BS found no polymorphisms in the coding regions of the ten genes but several SNPs in the shared promoter of one divergently transcribed GLP gene pair and a single SNP downstream of the transcribed region of a second GLP.ConclusionPhysical mapping and sequence comparison showed major haplotype divergence at the Sr2 locus between Hope and Chinese Spring. Candidate genes within the Sr2 region of Hope are being evaluated for the ability to confer stem rust resistance. Based on the detailed mapping and sequencing of the locus, we predict that Sr2 does not belong to the NB-LRR gene family and is not related to previously cloned, race non-specific rust resistance genes Lr34 and Yr36.
Building the sugarcane genome for biotechnology and identifying evolutionary trends.
BMC Genomics. 2014 Jun 30;15(1):540.
Voir plus
Authors
De Setta N, Monteiro-Vitorello CB, Metcalfe CJ, Cruz GM, Del Bem LE, Vicentini R, Nogueira FT, Campos RA, Nunes SL, Turrini PC, Vieira AP, Ochoa Cruz EA, Corrêa TC, Hotta CT, de Mello Varani A, Vautrin S, da Trindade AS, de Mendonça Vilela M, Lembke CG, Sato PM, de Andrade RF, Nishiyama MY Jr, Cardoso-Silva CB, Scortecci KC, Garcia AA, Carneiro MS, Kim C, Paterson AH, Bergès H, D Hont A, de Souza AP, Souza GM, Vincentz M, Kitajima JP, Van Sluys MA.
BMC Genomics. 2014 Jun 30;15(1):540.
Abstract
BACKGROUND:
Sugarcane is the source of sugar in all tropical and subtropical countries and is becoming increasingly important for bio-based fuels. However, its large (10 Gb), polyploid, complex genome has hindered genome based breeding efforts. Here we release the largest and most diverse set of sugarcane genome sequences to date, as part of an on-going initiative to provide a sugarcane genomic information resource, with the ultimate goal of producing a gold standard genome.
RESULTS:
Three hundred and seventeen chiefly euchromatic BACs were sequenced. A reference set of one thousand four hundred manually-annotated protein-coding genes was generated. A small RNA collection and a RNA-seq library were used to explore expression patterns and the sRNA landscape. In the sucrose and starch metabolism pathway, 16 non-redundant enzyme-encoding genes were identified. One of the sucrose pathway genes, sucrose-6-phosphate phosphohydrolase, is duplicated in sugarcane and sorghum, but not in rice and maize. A diversity analysis of the s6pp duplication region revealed haplotype-structured sequence composition. Examination of hom(e)ologous loci indicate both sequence structural and sRNA landscape variation. A synteny analysis shows that the sugarcane genome has expanded relative to the sorghum genome, largely due to the presence of transposable elements and uncharacterized intergenic and intronic sequences.
CONCLUSIONS:
This release of sugarcane genomic sequences will advance our understanding of sugarcane genetics and contribute to the development of molecular tools for breeding purposes and gene discovery.
Meiotic gene evolution: can you teach a new dog new tricks?
Mol Biol Evol. 2014
Voir plus
Authors
Lloyd A, Ranoux M, Vautrin S, Glover N, Fourment J, Charif D, Choulet F, Lassalle G, Marande W, Tran J, Granier F, Pingault L, Remay A, Marquis C, Belcram H, Chalhoub B, Feuillet C, Bergès H, Sourdille P, Jenczewski E.
MolBiol Evol. 2014
Abstract
Meiosis, the basis of sex, evolved through iterative gene duplications. To understand whether subsequent duplications have further enriched the core meiotic "tool-kit", we investigated the fate of meiotic gene duplicates following Whole Genome Duplication (WGD), a common occurrence in eukaryotes. We show that meiotic genes return to a single copy more rapidly than genome-wide average in Angiosperms, one of the lineages in which WGD is most vividly exemplified. The rate at which duplicates are lost decreases through time, a tendency that is also observed genome-wide and may thus prove to be a general trend post-WGD. The sharpest decline is observed for the subset of genes mediating meiotic recombination; however, we found no evidence that the presence of these duplicates is counter-selected in two recent polyploid crops selected for fertility. We therefore propose that their loss is passive, highlighting how quickly WGDs are resolved in the absence of selective duplicate retention.
The physical map of wheat chromosome 1BS provides insights into its gene space organization and evolution.
Genome Biol. 2013 Dec 20;14(12):R138.
Voir plus
Authors
Raats D, Frenkel Z, Krugman T, Dodek I, Sela H, Imková H, Magni F, Cattonaro F, Vautrin S, Bergès H, Wicker T, Keller B, Leroy P, Philippe R, Paux E, Dole El J, Feuillet C, Korol A, Fahima T
Genome Biol. 2013 Dec 20;14(12):R138.
Abstract
BACKGROUND:
The wheat genome sequence is an essential tool for advanced genomic research and improvements. The generation of a high-quality wheat genome sequence is challenging due to its complex 17 Gb polyploid genome. To overcome these difficulties, sequencing through the construction of BAC-based physical maps of individual chromosomes is employed by the wheat genomics community. Here, we present the construction of the first comprehensive physical map of chromosome 1BS, and illustrate its unique gene space organization and evolution.
RESULTS:
Fingerprinted BAC clones were assembled into 57 long scaffolds, anchored and ordered with 2,438 markers covering 83% of chromosome 1BS. The BAC-based chromosome 1BS physical map and gene order of the orthologous regions of model grass species were consistent, providing strong support for the reliability of the chromosome 1BS assembly. Chromosome 1BS gene space spans the entire length of the chromosome arm, with 76% of the genes organized in small gene-islands, accompanied by a two fold increase in gene density from the centromere to the telomere.
CONCLUSIONS:
This study provides new evidence on common and chromosome-specific features in the organization and evolution of the wheat genome, including a non-uniform distribution of gene density along the centromere-telomere axis, abundance of non-syntenic genes, the degree of colinearity with other grass genomes, and a non-uniform size expansion along the centromere-telomere axis, compared with other model cereal genomes. The high quality physical map constructed in this study provides a solid basis for the assembly of a reference sequence of chromosome 1BS and for breeding applications.
A Physical Map of the Short Arm of Wheat Chromosome 1A.
PLoS One. 8(11):e80272. doi: 10.1371/journal.pone.0080272.
Voir plus
Authors
Breen J, Wicker T, Shatalina M, Frenkel Z, Bertin I, Philippe R, Spielmeyer W, Simková H, Safář J, Cattonaro F, Scalabrin S, Magni F, Vautrin S, Bergès H; International Wheat Genome Sequencing Consortium, Paux E, Fahima T, Doležel J, Korol A, Feuillet C, Keller B.
PLoS One. 8(11):e80272. doi: 10.1371/journal.pone.0080272.
Abstract
Bread wheat (Triticum aestivum) has a large and highly repetitive genome which poses major technical challenges for its study. To aid map-based cloning and future genome sequencing projects, we constructed a BAC-based physical map of the short arm of wheat chromosome 1A (1AS). From the assembly of 25,918 high information content (HICF) fingerprints from a 1AS-specific BAC library, 715 physical contigs were produced that cover almost 99% of the estimated size of the chromosome arm. The 3,414 BAC clones constituting the minimum tiling path were end-sequenced. Using a gene microarray containing ∼40 K NCBI UniGene EST clusters, PCR marker screening and BAC end sequences, we arranged 160 physical contigs (97 Mb or 35.3% of the chromosome arm) in a virtual order based on synteny with Brachypodium, rice and sorghum. BAC end sequences and information from microarray hybridisation was used to anchor 3.8 Mbp of Illumina sequences from flow-sorted chromosome 1AS to BAC contigs. Comparison of genetic and synteny-based physical maps indicated that ∼50% of all genetic recombination is confined to 14% of the physical length of the chromosome arm in the distal region. The 1AS physical map provides a framework for future genetic mapping projects as well as the basis for complete sequencing of chromosome arm 1AS.
A high density physical map of chromosome 1BL supports evolutionary studies, map-based cloning and sequencing in wheat.
Genome Biol. 2013 Jun 25;14(6):R64. [Epub ahead of print]
Voir plus
Authors
Philippe R, Paux E, Bertin I, Sourdille P, Choulet F, Laugier C, Simkova H, Safar J, Bellec A, Vautrin S, Frenkel Z, Cattonaro F, Magni F, Scalabrin S, Martis MM, Mayer KF, Korol A, Berges H, Dolezel J, Feuillet C.
Genome Biol. 2013 Jun 25;14(6):R64. [Epub ahead of print]
Abstract
BACKGROUND:
As for other major crops, achieving a complete wheat genome sequence is essential for the application of genomics to breeding new and improved varieties. To overcome the complexities of the large, highly repetitive and hexaploid wheat genome, the International Wheat Genome Sequencing Consortium established a chromosome-based strategy that was validated by the construction of the physical map of chromosome 3B. Here, we present improved strategies for the construction of highly integrated and ordered wheat physical maps, using chromosome 1BL as a template, and illustrate their potential for evolutionary studies and map-based cloning.
RESULTS:
Using a combination of novel high throughput marker assays and an assembly program, we developed a high quality physical map representing 93% of wheat chromosome 1BL, anchored and ordered with 5,489 markers including 1,161 genes. Analysis of the gene space organization and evolution revealed that gene distribution and conservation along the chromosome results from the superimposition of the ancestral grass and recent wheat evolutionary patterns leading to a peak of synteny in the central part of the chromosome arm and an increased density of non collinear genes towards the telomere. With a density of about 11 markers per Mb, the 1BL physical map provides 916 markers, including 193 genes, for fine mapping the 40 QTLs mapped on this chromosome.
CONCLUSIONS:
Here, we demonstrate that high marker density physical maps can be developed in complex genomes such as wheat to accelerate map-based cloning, gain new insights into genome evolution, and provide a foundation for reference sequencing.:
Physical Mapping Integrated with Syntenic Analysis to Characterize the Gene Space of the Long Arm of Wheat Chromosome 1A.
PLoS One. 2013 Apr 16;8(4):e59542. doi: 10.1371/journal.pone.0059542.
Voir plus
Authors
Lucas SJ, Akpınar BA, Kantar M, Weinstein Z, Aydınoğlu F, Safář J, Simková H, Frenkel Z, Korol A, Magni F, Cattonaro F, Vautrin S, Bellec A, Bergès H, Doležel J, Budak H.
PLoS One. 2013 Apr 16;8(4):e59542. doi: 10.1371/journal.pone.0059542.
Abstract
BACKGROUND:
Bread wheat (Triticum aestivum L.) is one of the most important crops worldwide and its production faces pressing challenges, the solution of which demands genome information. However, the large, highly repetitive hexaploid wheat genome has been considered intractable to standard sequencing approaches. Therefore the International Wheat Genome Sequencing Consortium (IWGSC) proposes to map and sequence the genome on a chromosome-by-chromosome basis.
METHODOLOGY PRINCIPAL FINDINGS:
We have constructed a physical map of the long arm of bread wheat chromosome 1A using chromosome-specific BAC libraries by High Information Content Fingerprinting (HICF). Two alternative methods (FPC and LTC) were used to assemble the fingerprints into a high-resolution physical map of the chromosome arm. A total of 365 molecular markers were added to the map, in addition to 1122 putative unique transcripts that were identified by microarray hybridization. The final map consists of 1180 FPC-based or 583 LTC-based contigs.
CONCLUSIONS SIGNIFICANCE:
The physical map presented here marks an important step forward in mapping of hexaploid bread wheat. The map is orders of magnitude more detailed than previously available maps of this chromosome, and the assignment of over a thousand putative expressed gene sequences to specific map locations will greatly assist future functional studies. This map will be an essential tool for future sequencing of and positional cloning within chromosome 1A.
A 3000-loci transcription map of chromosome 3B unravels the structural and functional features of gene islands in hexaploid wheat.
Plant Physiol. 2011 Oct 27.
Voir plus
Authors
Rustenholz C, Choulet F, Laugier C, Safár J, Simková H, Dolezel J, Magni F, Scalabrin S, Cattonaro F, Vautrin S, Bellec A, Bergès H, Feuillet C, Paux E.
Plant Physiol. 2011 Oct 27.
Abstract
To improve our understanding of the organization and regulation of the wheat (Triticum aestivum L.) gene space, we established the first transcription map of a wheat chromosome (3B) by hybridizing a newly developed wheat expression microarray with BAC pools from a new version of the 3B physical map as well as with cDNA probes derived from 15 RNA samples. Mapping data for almost 3000 genes showed that the gene space spans the whole chromosome 3B with a twofold increase of gene density towards the telomeres due to an increase in the number of genes in islands. Comparative analyses with rice and Brachypodium revealed that these gene islands are composed mainly of genes likely originating from interchromosomal gene duplications. Gene ontology and expression profile analyses for the 3000 genes located along the chromosome revealed that the gene islands are enriched significantly in genes sharing the same function or expression profile, thereby suggesting that genes in islands acquired shared regulation during evolution. Only a small fraction of these clusters of cofunctional and coexpressed genes was conserved with rice and Brachypodium indicating a recent origin. Finally, genes with the same expression profiles in remote islands (coregulation islands) were identified suggesting long-distance regulation of gene expression along the chromosomes in wheat.
Functional features of a single chromosome arm in wheat (1AL) determined from its structure.
Funct Integr Genomics. 2011 Sep 3.
Voir plus
Authors :
Lucas SJ, Simková H, Safár J, Jurman I, Cattonaro F, Vautrin S, Bellec A, Berges H, Doležel J, Budak H.
Funct Integr Genomics. 2011 Sep 3.
Abstract :
Bread wheat (Triticum aestivum L.) is one of the most important crops globally and a high priority for genetic improvement, but its large and complex genome has been seen as intractable to whole genome sequencing. Isolation of individual wheat chromosome arms has facilitated large-scale sequence analyses. However, so far there is no such survey of sequences from the A genome of wheat. Greater understanding of an A chromosome could facilitate wheat improvement and future sequencing of the entire genome. We have constructed BAC library from the long arm of T. aestivum chromosome 1A (1AL) and obtained BAC end sequences from 7,470 clones encompassing the arm. We obtained 13,445 (89.99%) useful sequences with a cumulative length of
7.57 Mb, representing 1.43% of 1AL and about 0.14% of the entire A genome. The GC content of the sequences was 44.7%, and 90% of the chromosome was estimated to comprise repeat sequences, while just over 1% encoded expressed genes. From the sequence data, we identified a large number of sites suitable for development of molecular markers
(362 SSR and 6,948 ISBP) which will have utility for mapping this chromosome and for marker assisted breeding. From 44 putative ISBP markers tested 23 (52.3%) were found to be useful. The BAC end sequence data also enabled the identification of genes and syntenic blocks specific to chromosome 1AL, suggesting regions of particular functional interest and targets for future research.
Advancing Eucalyptus genomics: identification and sequencing of lignin biosynthesis genes from deep-coverage BAC libraries
BMC Genomics. 2011 Mar 4;12(1):137.
Voir plus
Authors :
Paiva JA, Prat E, Vautrin S, Santos MD, San-Clemente H, Brommonschenkel S, Fonseca PG, Grattapaglia D, Song X, Ammiraju JS, Kudrna D, Wing RA, Freitas AT, Berges H, Grima-Pettenati J.
BMC Genomics. 2011 Mar 4;12(1):137.
Abstract :
BACKGROUND: Eucalyptus species are among the most planted hardwoods in the world because of their rapid growth, adaptability and valuable wood properties. The development and integration of genomic resources into breeding practice will be increasingly important in the decades to come. Bacterial artificial chromosome (BAC) libraries are key genomic tools that enable positional cloning of important traits, synteny evaluation, and the development of genome framework physical maps for genetic linkage and genome sequencing.
RESULTS: We describe the construction and characterization of two deep-coverage BAC libraries EG_Ba and EG_Bb obtained from nuclear DNA fragments of E. grandis (clone BRASUZ1) digested with HindIII and BstYI, respectively. Genome coverages of 17 and 15 haploid genome equivalents were estimated for EG_Ba and EG_Bb, respectively. Both libraries contained large inserts, with average sizes ranging from 135Kb (Eg_Bb) to 157Kb (Eg_Ba), very low extra-nuclear genome contamination providing a probability of finding a single copy gene [greater than or equal to] 99.99%. Libraries were screened for the presence of several genes of interest via hybridizations to high-density BAC filters followed by PCR validation. Five selected BAC clones were sequenced and assembled using the Roche GS FLX technology providing the whole sequence of the E. grandis chloroplast genome, and complete genomic sequences of important lignin biosynthesis genes.
CONCLUSIONS: The two E. grandis BAC libraries described in this study represent an important milestone for the advancement of Eucalyptus genomics and forest tree research. These BAC resources have a highly redundant genome coverage (>15x), contain large average inserts and have a very low percentage of clones with organellar DNA or empty vectors. These publicly available BAC libraries are thus suitable for a broad range of applications in genetic and genomic research in Eucalyptus and possibly in related species of Myrtaceae, including genome sequencing, gene isolation, functional and comparative genomics. Because they have been constructed using the same tree (E. grandis BRASUZ1) whose full genome is being sequenced, they should prove instrumental for assembly and gap filling of the upcoming Eucalyptus reference genome sequence.
Brassica orthologs from BANYULS belong to a small multigene family, which is involved in procyanidin accumulation in the seed
Planta. 2009 Sep 17.
Voir plus
Abstract
As part of a research programme focused on flavonoid biosynthesis in the seed coat of Brassica napus L. (oilseed rape), orthologs of the BANYULS gene that encoded anthocyanidin reductase were cloned in B. napus as well as in the related species Brassica rapa and Brassica oleracea. B. napus genome contained four functional copies of BAN, two originating from each diploid progenitor. Amino acid sequences were highly conserved between the Brassicaceae including B. napus, B. rapa, B. oleracea as well as the model plant Arabidopsis thaliana. Along the 200 bp in 5' of the ATG codon, Bna.BAN promoters (ProBna.BAN) were conserved with AtANR promoter and contained putative cis-acting elements. In addition, transgenic Arabidopsis and oilseed rape plants carrying the first 230 bp of ProBna.BAN fused to the UidA reporter gene were generated. In the two Brassicaceae backgrounds, ProBna.BAN activity was restricted to the seed coat. In B. napus seed, ProBna.BAN was activated in procyanidin-accumulating cells, namely the innermost layer of the inner integument and the micropyle-chalaza area. At the transcriptional level, the four Bna.BAN genes were expressed in the seed. Laser microdissection assays of the seed integuments showed that Bna.BAN expression was restricted to the inner integument, which was consistent with the activation profile of ProBna.BAN. Finally, Bna.BAN genes were mapped onto oilseed rape genetic maps and potential co-localisations with seed colour quantitative trait loci are discussed.
Planta. 2009 Sep 17 - Auger et al.
Authors
Bathilde Auger, Cécile Baron, Marie-Odile Lucas, Sonia Vautrin, Hélène Bergès, Boulos Chalhoub, Alain Fautrel, Michel Renard, Nathalie Nesi.
Planta. 2009 Sep 17.
Fine Mapping and Marker Development for the Crossability Gene SKr on Chromosome 5BS of Hexaploid Wheat (Triticum aestivum L.)
Genetics. 2009 Aug 3.
Voir plus
Abstract
Most elite wheat varieties cannot be crossed with related species thereby restricting greatly the germplasm that can be used for alien introgression in breeding programs. Inhibition to crossability is controlled genetically and a number of QTL have been identified to date, including SKr, a strong QTL affecting crossability between wheat and rye located at the distal end of chromosome 5BS. In this study, we used a recombinant SSD population originating from a cross between the poorly crossable cultivar Courtot and the crossable line MP98 to characterize the major dominant effect of SKr and map the gene at the distal end of the chromosome near the 5B homoeologous GSP locus. Colinearity with barley and rice was used to saturate the SKr region with new markers and establish orthologous relationships with a 52 kb region on rice chromosome 12. In total, five markers were mapped within a genetic interval of 0.3 cM and 400 kb of BAC contigs were established on both sides of the gene to lay the foundation for map-based cloning of SKr. Two SSR markers completely linked to SKr were used to evaluate a collection of crossable wheat progenies originating from primary triticale breeding programs. The results confirm the major effect of SKr on crossability and the usefulness of the two markers for the efficient introgression of crossability in elite wheat varieties.
Authors
Alfares W, Bouguennec A, Balfourier F, Gay G, Bergès H, Vautrin S, Sourdille P, Bernard M, Feuillet C.
Genetics. 2009 Aug 3.